

Towards population-scale long-read sequencing
- PMID: 34050336
- PMCID: PMC8161719
- DOI: 10.1038/s41576-021-00367-3
Towards population-scale long-read sequencing
Abstract
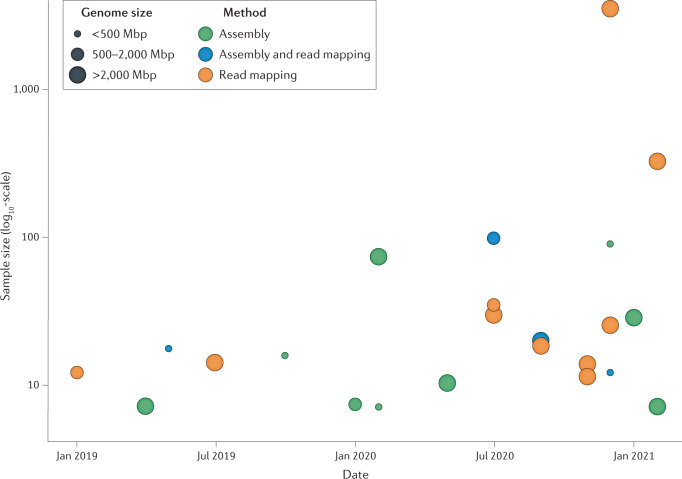
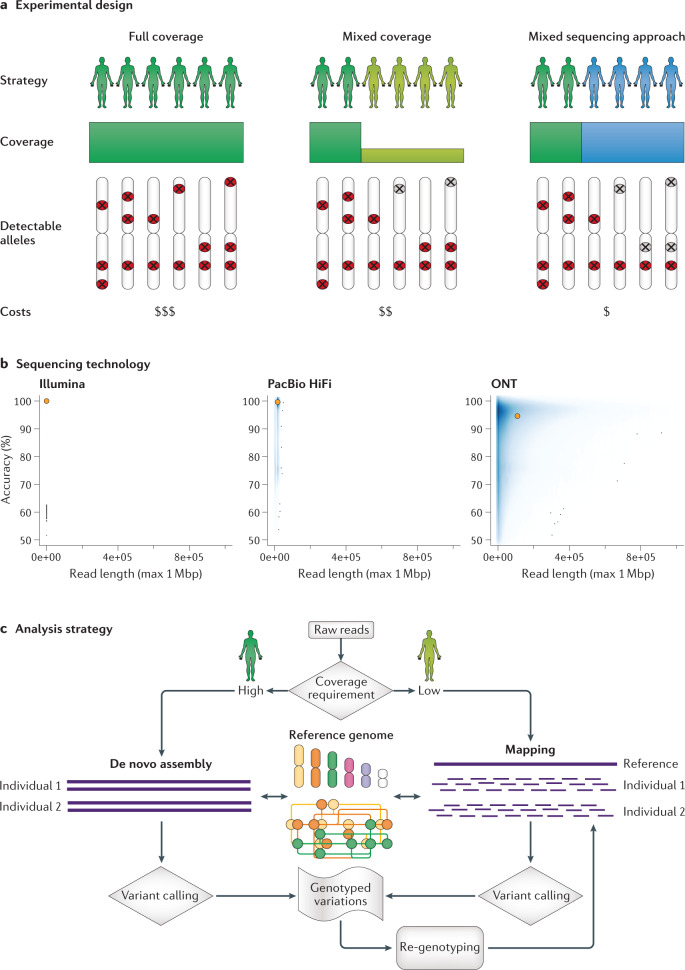
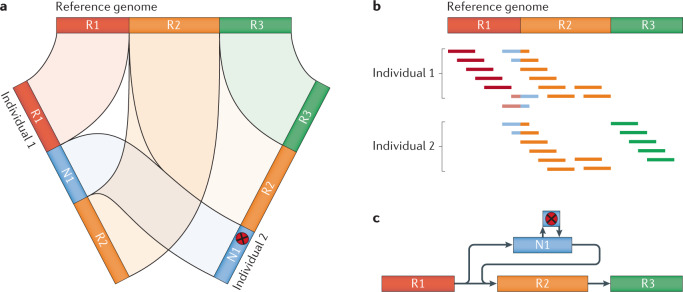
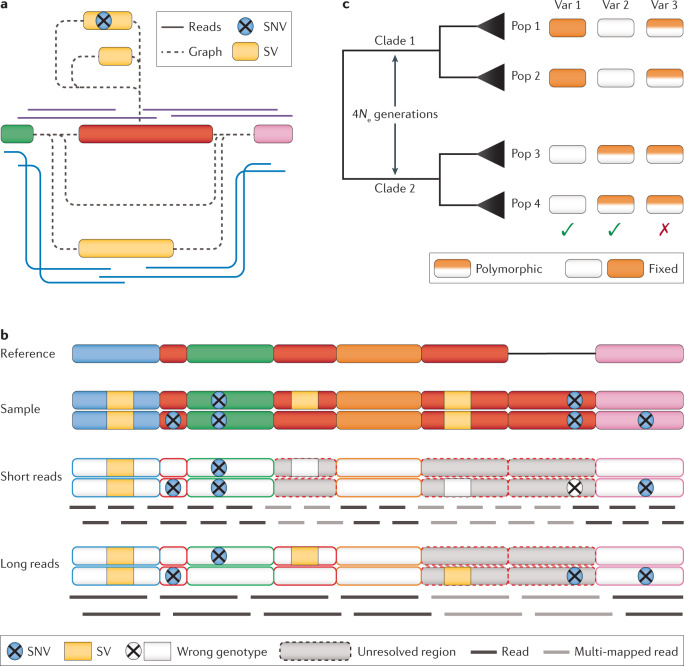
Long-read sequencing technologies have now reached a level of accuracy and yield that allows their application to variant detection at a scale of tens to thousands of samples. Concomitant with the development of new computational tools, the first population-scale studies involving long-read sequencing have emerged over the past 2 years and, given the continuous advancement of the field, many more are likely to follow. In this Review, we survey recent developments in population-scale long-read sequencing, highlight potential challenges of a scaled-up approach and provide guidance regarding experimental design. We provide an overview of current long-read sequencing platforms, variant calling methodologies and approaches for de novo assemblies and reference-based mapping approaches. Furthermore, we summarize strategies for variant validation, genotyping and predicting functional impact and emphasize challenges remaining in achieving long-read sequencing at a population scale.
© 2021. Springer Nature Limited.
Conflict of interest statement
W.D.C. and F.J.S. have received sponsored travel from PacBio and/or Oxford Nanopore. M.H.W. declares no competing interests.
Figures
References
-
- Hartman KA, Rashkin SR, Witte JS, Hernandez RD. Imputed genomic data reveals a moderate effect of low frequency variants to the heritability of complex human traits. bioRxiv. 2019 doi: 10.1101/2019.12.18.879916. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
