

Small molecules in targeted cancer therapy: advances, challenges, and future perspectives
- PMID: 34054126
- PMCID: PMC8165101
- DOI: 10.1038/s41392-021-00572-w
Small molecules in targeted cancer therapy: advances, challenges, and future perspectives
Abstract
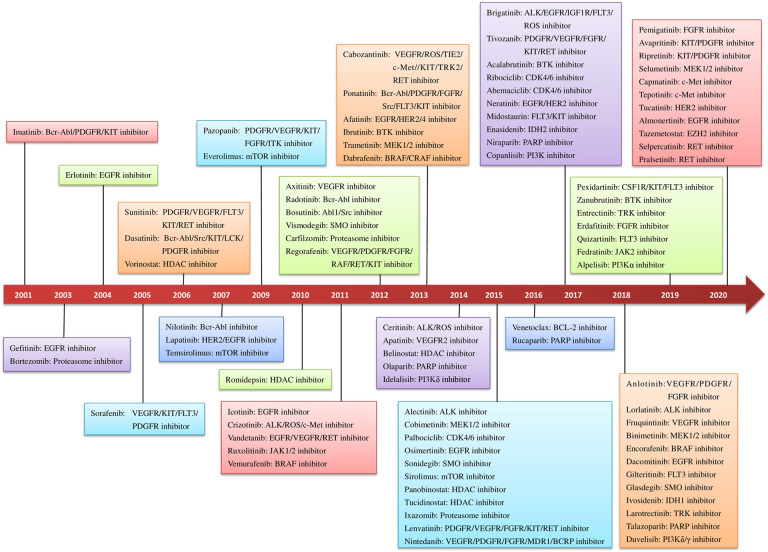
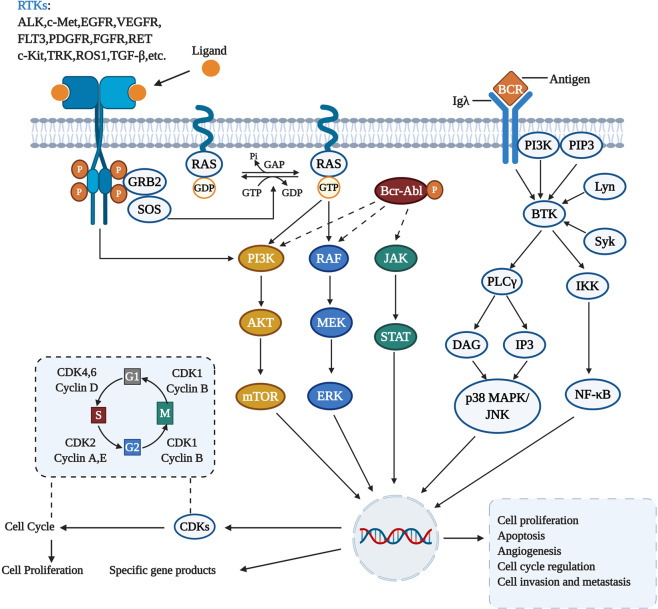
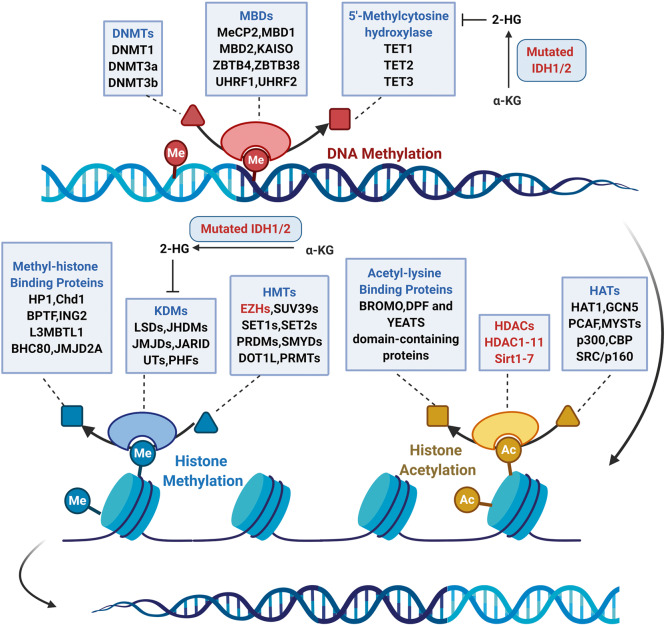
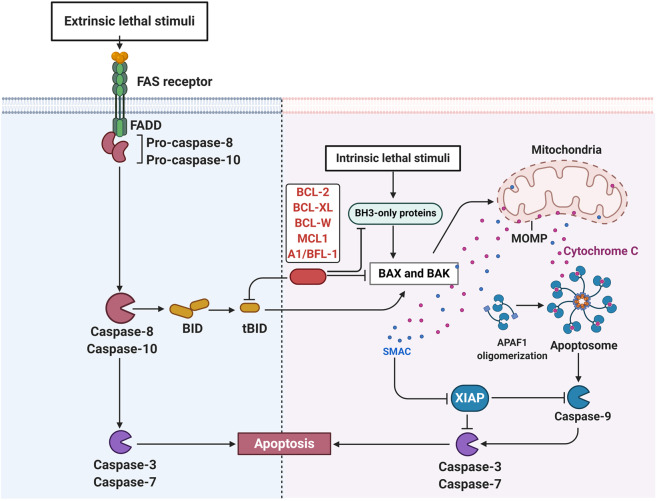
Due to the advantages in efficacy and safety compared with traditional chemotherapy drugs, targeted therapeutic drugs have become mainstream cancer treatments. Since the first tyrosine kinase inhibitor imatinib was approved to enter the market by the US Food and Drug Administration (FDA) in 2001, an increasing number of small-molecule targeted drugs have been developed for the treatment of malignancies. By December 2020, 89 small-molecule targeted antitumor drugs have been approved by the US FDA and the National Medical Products Administration (NMPA) of China. Despite great progress, small-molecule targeted anti-cancer drugs still face many challenges, such as a low response rate and drug resistance. To better promote the development of targeted anti-cancer drugs, we conducted a comprehensive review of small-molecule targeted anti-cancer drugs according to the target classification. We present all the approved drugs as well as important drug candidates in clinical trials for each target, discuss the current challenges, and provide insights and perspectives for the research and development of anti-cancer drugs.
Conflict of interest statement
Shengyong Yang is the editorial board member of Signal Transduction and Targeted Therapy, but he has not been involved in the process of manuscript handling. The remaining authors declare no competing interests.
Figures
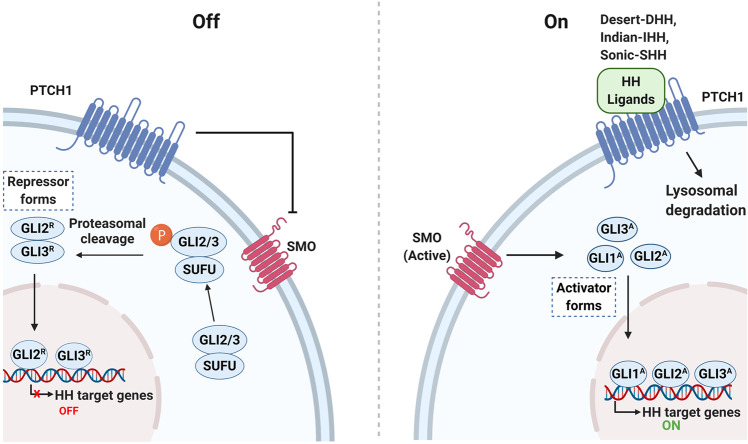
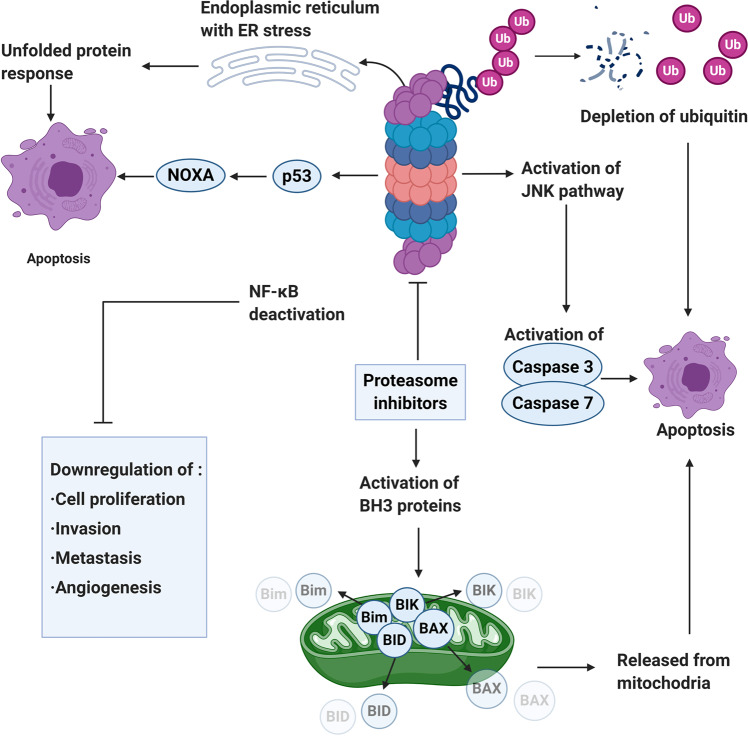
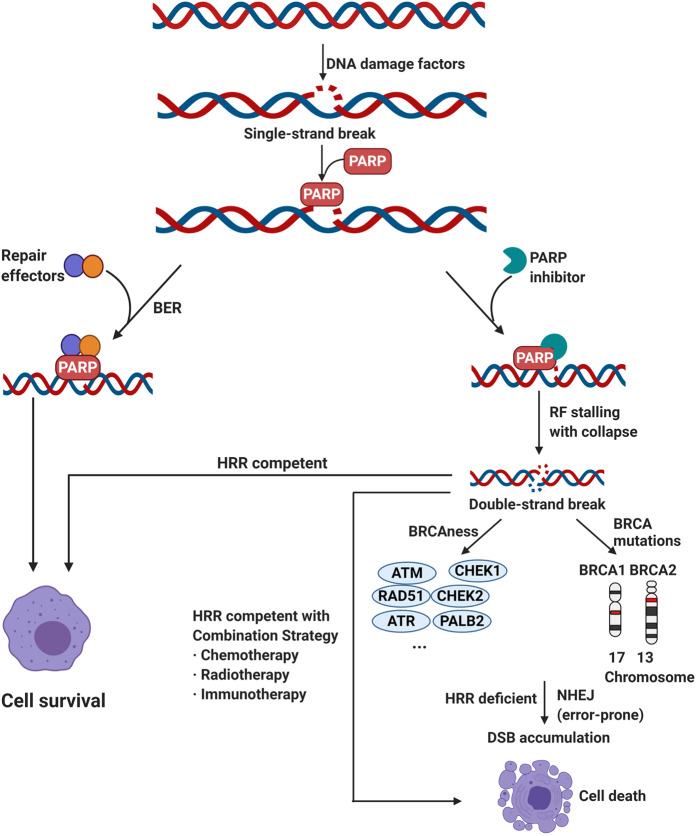
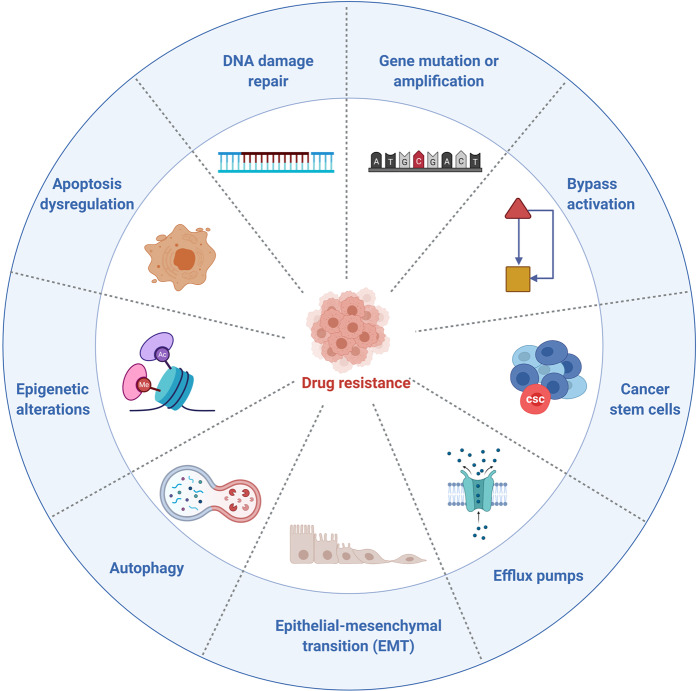
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
