

Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments
- PMID: 34055195
- PMCID: PMC8149218
- DOI: 10.1155/2021/6614009
Role of Oxidative Stress in Reperfusion following Myocardial Ischemia and Its Treatments
Abstract
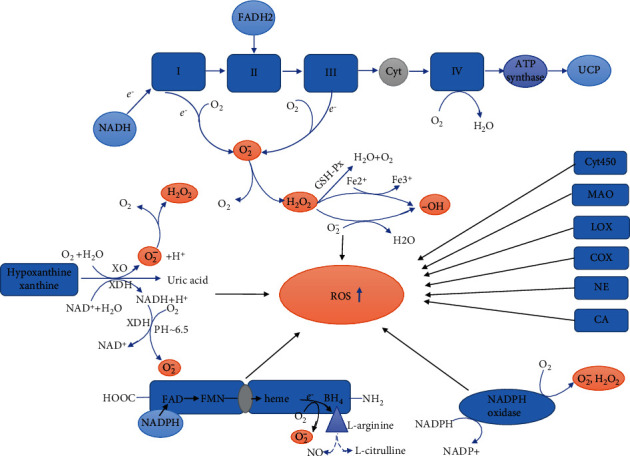
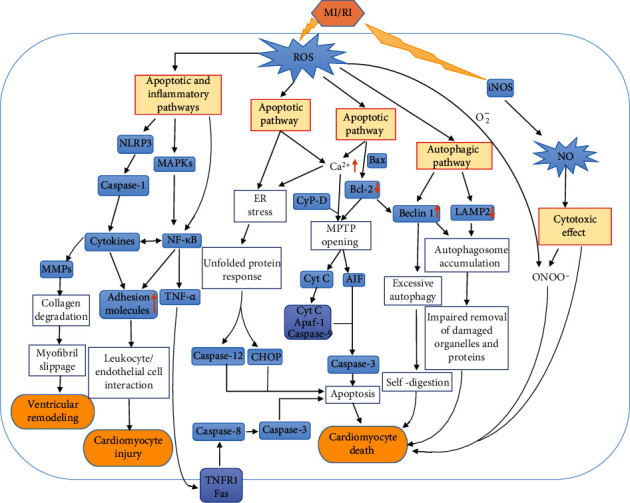
Myocardial ischemia is a disease with high morbidity and mortality, for which reperfusion is currently the standard intervention. However, the reperfusion may lead to further myocardial damage, known as myocardial ischemia/reperfusion injury (MI/RI). Oxidative stress is one of the most important pathological mechanisms in reperfusion injury, which causes apoptosis, autophagy, inflammation, and some other damage in cardiomyocytes through multiple pathways, thus causing irreversible cardiomyocyte damage and cardiac dysfunction. This article reviews the pathological mechanisms of oxidative stress involved in reperfusion injury and the interventions for different pathways and targets, so as to form systematic treatments for oxidative stress-induced myocardial reperfusion injury and make up for the lack of monotherapy.
Copyright © 2021 Mi Xiang et al.
Conflict of interest statement
The authors declare that they do not have anything to disclose regarding conflict of interest with respect to this manuscript.
Figures
Similar articles
-
[Mechanisms of oxidative stress in myocardial ischemia-reperfusion injury and protective effects of traditional Chinese medicines].Zhongguo Zhong Yao Za Zhi. 2024 Jul;49(13):3452-3461. doi: 10.19540/j.cnki.cjcmm.20240312.701. Zhongguo Zhong Yao Za Zhi. 2024. PMID: 39041117 Review. Chinese.
-
Crosstalk among Reactive Oxygen Species, Autophagy and Metabolism in Myocardial Ischemia and Reperfusion Stages.Aging Dis. 2024 May 7;15(3):1075-1107. doi: 10.14336/AD.2023.0823-4. Aging Dis. 2024. PMID: 37728583 Free PMC article. Review.
-
MiR-302a-3p aggravates myocardial ischemia-reperfusion injury by suppressing mitophagy via targeting FOXO3.Exp Mol Pathol. 2020 Dec;117:104522. doi: 10.1016/j.yexmp.2020.104522. Epub 2020 Aug 29. Exp Mol Pathol. 2020. PMID: 32866521
-
Reduced silent information regulator 1 signaling exacerbates myocardial ischemia-reperfusion injury in type 2 diabetic rats and the protective effect of melatonin.J Pineal Res. 2015 Oct;59(3):376-90. doi: 10.1111/jpi.12269. Epub 2015 Sep 11. J Pineal Res. 2015. PMID: 26327197
-
Icaritin Attenuates Myocardial Ischemia and Reperfusion Injury Via Anti-Inflammatory and Anti-Oxidative Stress Effects in Rats.Am J Chin Med. 2015;43(6):1083-97. doi: 10.1142/S0192415X15500627. Epub 2015 Sep 14. Am J Chin Med. 2015. PMID: 26364662
Cited by
-
miR-124-3p downregulates EGR1 to suppress ischemia-hypoxia reperfusion injury in human iPS cell-derived cardiomyocytes.Sci Rep. 2024 Jun 27;14(1):14811. doi: 10.1038/s41598-024-65373-x. Sci Rep. 2024. PMID: 38926457 Free PMC article.
-
Tapping into Nature's Arsenal: Harnessing the Potential of Natural Antioxidants for Human Health and Disease Prevention.Foods. 2024 Jun 25;13(13):1999. doi: 10.3390/foods13131999. Foods. 2024. PMID: 38998505 Free PMC article. Review.
-
Targeting oxidative stress as a preventive and therapeutic approach for cardiovascular disease.J Transl Med. 2023 Aug 2;21(1):519. doi: 10.1186/s12967-023-04361-7. J Transl Med. 2023. PMID: 37533007 Free PMC article. Review.
-
Effect of PCI on ophthalmic artery hemodynamics in patients with acute coronary syndrome.Front Med (Lausanne). 2024 Mar 4;11:1367900. doi: 10.3389/fmed.2024.1367900. eCollection 2024. Front Med (Lausanne). 2024. PMID: 38500953 Free PMC article.
-
Ferroptosis in organ ischemia-reperfusion injuries: recent advancements and strategies.Mol Cell Biochem. 2025 Jan;480(1):19-41. doi: 10.1007/s11010-024-04978-2. Epub 2024 Mar 31. Mol Cell Biochem. 2025. PMID: 38556592 Review.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
