

De Novo Sequencing Provides Insights Into the Pathogenicity of Foodborne Vibrio parahaemolyticus
- PMID: 34055666
- PMCID: PMC8162212
- DOI: 10.3389/fcimb.2021.652957
De Novo Sequencing Provides Insights Into the Pathogenicity of Foodborne Vibrio parahaemolyticus
Abstract
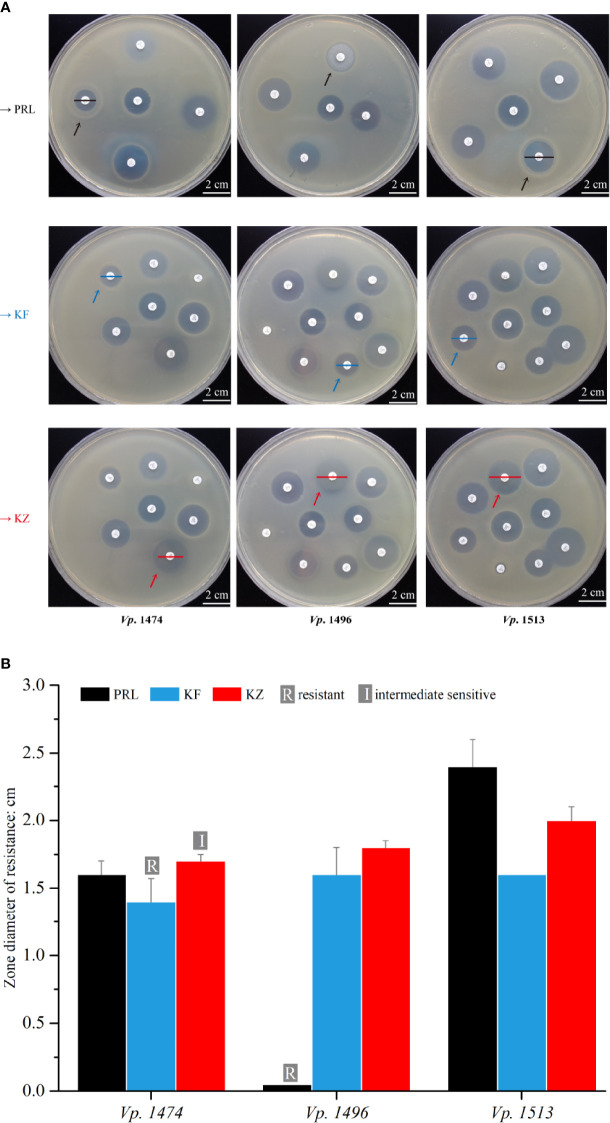
Vibrio parahaemolyticus is a common pathogenic marine bacterium that causes gastrointestinal infections and other health complications, which could be life-threatening to immunocompromised patients. For the past two decades, the pathogenicity of environmental V. parahaemolyticus has increased greatly, and the genomic change behind this phenomenon still needs an in-depth exploration. To investigate the difference in pathogenicity at the genomic level, three strains with different hemolysin expression and biofilm formation capacity were screened out of 69 environmental V. parahaemolyticus strains. Subsequently, 16S rDNA analysis, de novo sequencing, pathogenicity test, and antibiotic resistance assays were performed. Comparative genome-scale interpretation showed that various functional region differences in pathogenicity of the selected V. parahaemolyticus strains were due to dissimilarities in the distribution of key genetic elements and in the secretory system compositions. Furthermore, the genomic analysis-based hypothesis of distinct pathogenic effects was verified by the survival rate of mouse models infected with different V. parahaemolyticus strains. Antibiotic resistance results also presented the multi-directional evolutionary potential in environmental V. parahaemolyticus, in agreement with the phylogenetic analysis results. Our study provides a theoretical basis for better understanding of the increasing pathogenicity of environmental V. parahaemolyticus at the genome level. Further, it has a key referential value for the exploration of pathogenicity and prevention of environmental V. parahaemolyticus in the future.
Keywords: Vibrio parahaemolyticus; de novo sequencing; mouse model; pathogenicity; virulence.
Copyright © 2021 Liu, Qin, Wu, Fu, Yu and Zhou.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








Similar articles
-
Trh (tdh-/trh+) gene analysis of clinical, environmental and food isolates of Vibrio parahaemolyticus as a tool for investigating pathogenicity.Int J Food Microbiol. 2016 May 16;225:43-53. doi: 10.1016/j.ijfoodmicro.2016.02.016. Epub 2016 Mar 2. Int J Food Microbiol. 2016. PMID: 26990408
-
Comparative genomic analysis of clinical and environmental strains provides insight into the pathogenicity and evolution of Vibrio parahaemolyticus.BMC Genomics. 2014 Dec 18;15(1):1135. doi: 10.1186/1471-2164-15-1135. BMC Genomics. 2014. PMID: 25518728 Free PMC article.
-
Distribution of genes encoding four pathogenicity islands (VPaIs), T6SS, biofilm, and type I pilus in food and clinical strains of Vibrio parahaemolyticus in China.Foodborne Pathog Dis. 2010 Jun;7(6):649-58. doi: 10.1089/fpd.2009.0441. Foodborne Pathog Dis. 2010. PMID: 20132020
-
Ecological fitness and virulence features of Vibrio parahaemolyticus in estuarine environments.Appl Microbiol Biotechnol. 2017 Mar;101(5):1781-1794. doi: 10.1007/s00253-017-8096-9. Epub 2017 Jan 31. Appl Microbiol Biotechnol. 2017. PMID: 28144705 Review.
-
Epidemiology, pathogenesis, and prevention of foodborne Vibrio parahaemolyticus infections.Foodborne Pathog Dis. 2004 Summer;1(2):74-88. doi: 10.1089/153531404323143594. Foodborne Pathog Dis. 2004. PMID: 15992266 Review.
Cited by
-
Biological and transcriptional studies reveal VmeL is involved in motility, biofilm formation and virulence in Vibrio parahaemolyticus.Front Microbiol. 2022 Aug 9;13:976334. doi: 10.3389/fmicb.2022.976334. eCollection 2022. Front Microbiol. 2022. PMID: 36016795 Free PMC article.
-
Isolation of the Thermostable β-Glucosidase-Secreting Strain Bacillus altitudinis JYY-02 and Its Application in the Production of Gardenia Blue.Microbiol Spectr. 2022 Aug 31;10(4):e0153522. doi: 10.1128/spectrum.01535-22. Epub 2022 Jul 14. Microbiol Spectr. 2022. PMID: 35863007 Free PMC article.
-
Identification of Antibacterial Components and Modes in the Methanol-Phase Extract from a Herbal Plant Potentilla kleiniana Wight et Arn.Foods. 2023 Apr 13;12(8):1640. doi: 10.3390/foods12081640. Foods. 2023. PMID: 37107435 Free PMC article.
References
-
- Akhter S., McNair K., Decewicz P. (2019). "PhiSpy". 3.4 ed (Cambridge, Massachusetts, United States: Massachusetts Institute of Technology; ).
-
- Centers for Disease Control and Prevention (CDC) (2019). Surveillance for Foodborne Disease Outbreaks, United States. Annual Report. (2017). (Atlanta, Georgia: U.S. Department of Health and Human Services; ), 3–10.
-
- Baker-Austin C., Trinanes J. A., Taylor N. G. H., Hartnell R., Siitonen A., Martinez-Urtaza J. (2013). Emerging Vibrio risk at high latitudes in response to ocean warming. Nat. Clim. Chang. 31 (1), 73–77. 10.1038/nclimate1628 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous