

Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis
- PMID: 34059824
- PMCID: PMC9309980
- DOI: 10.1038/s41591-021-01346-1
Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis
Abstract
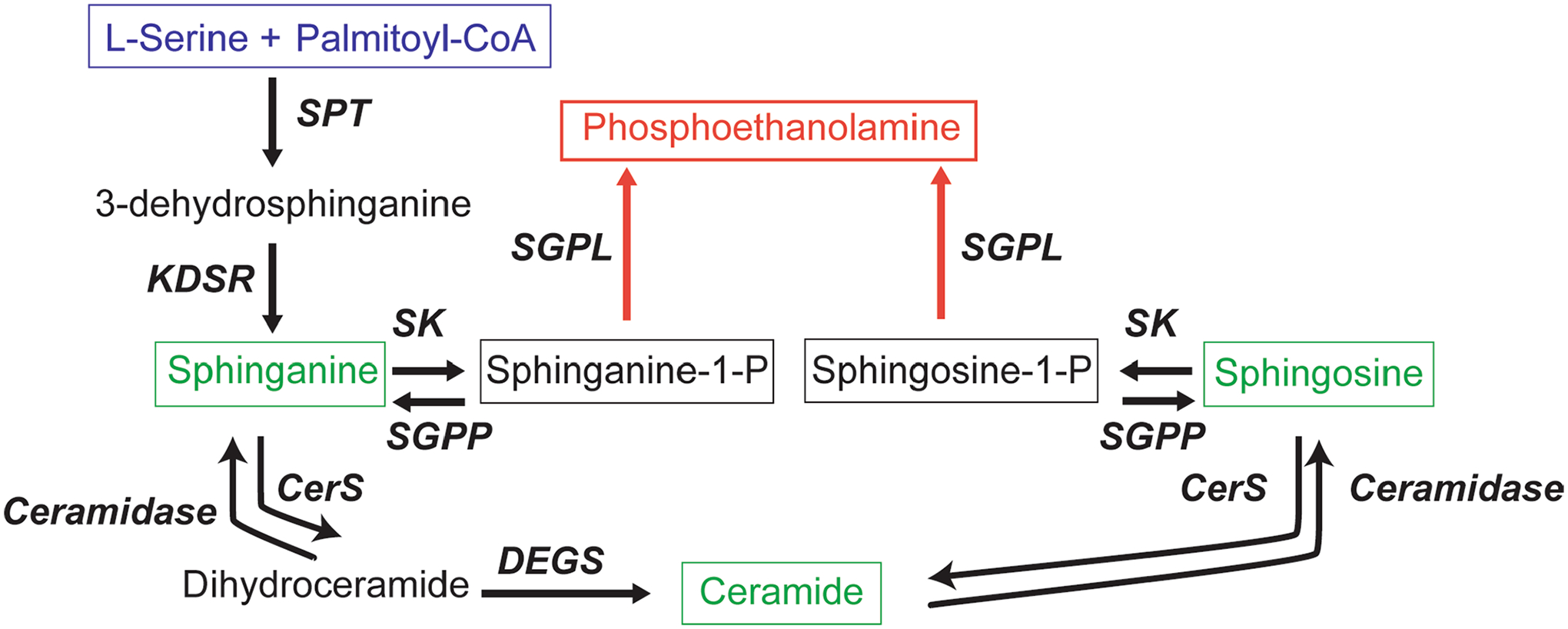
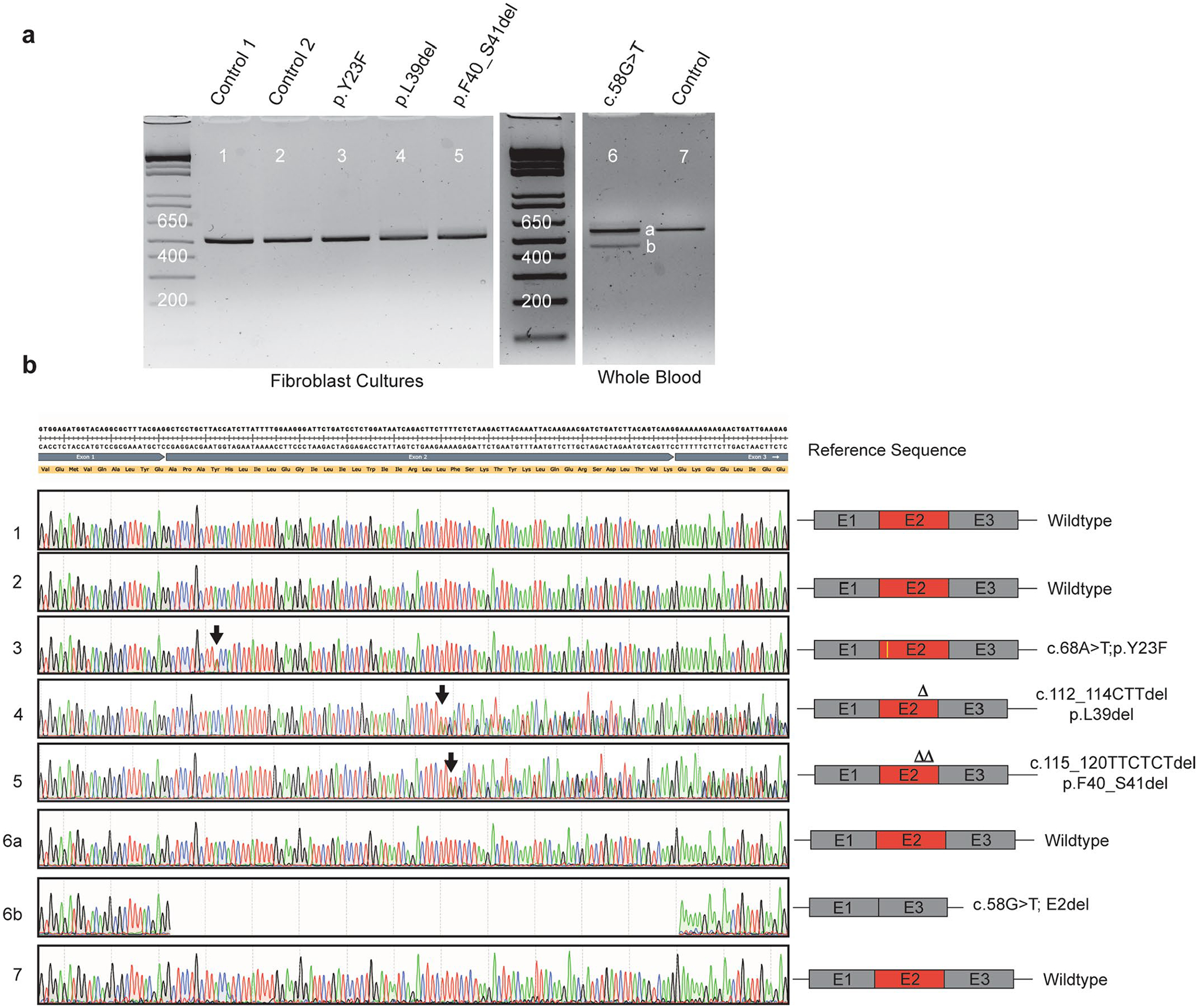
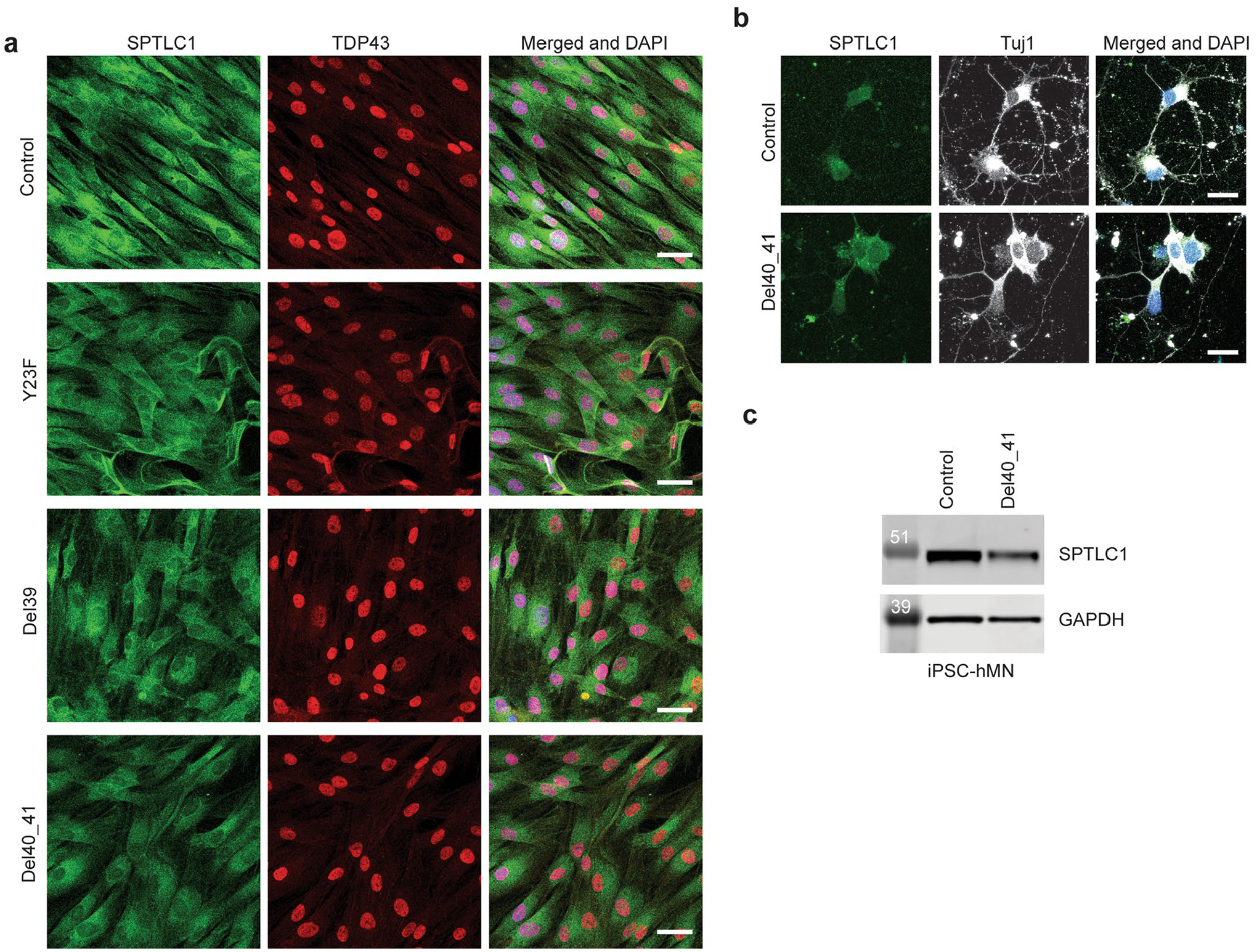
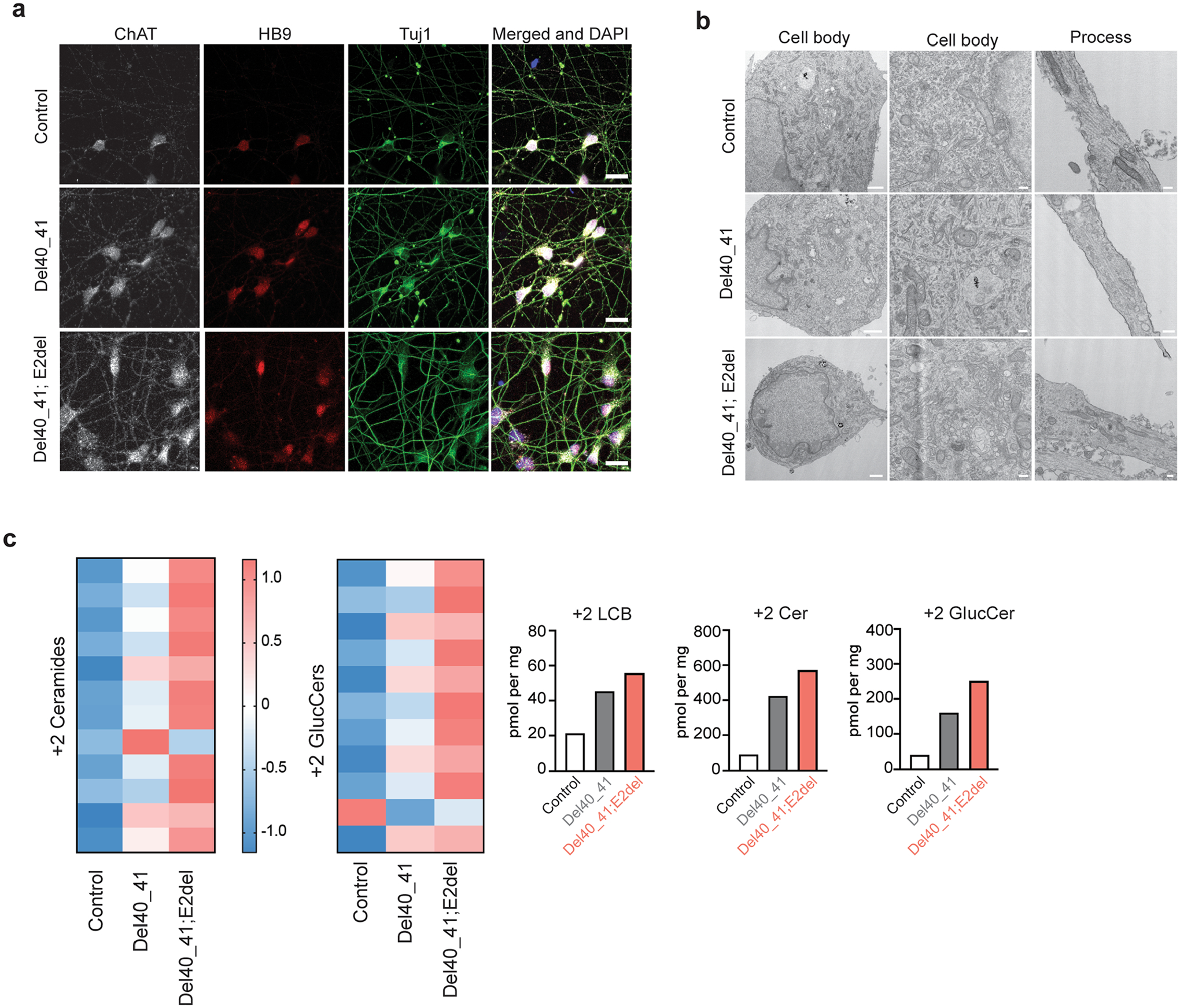
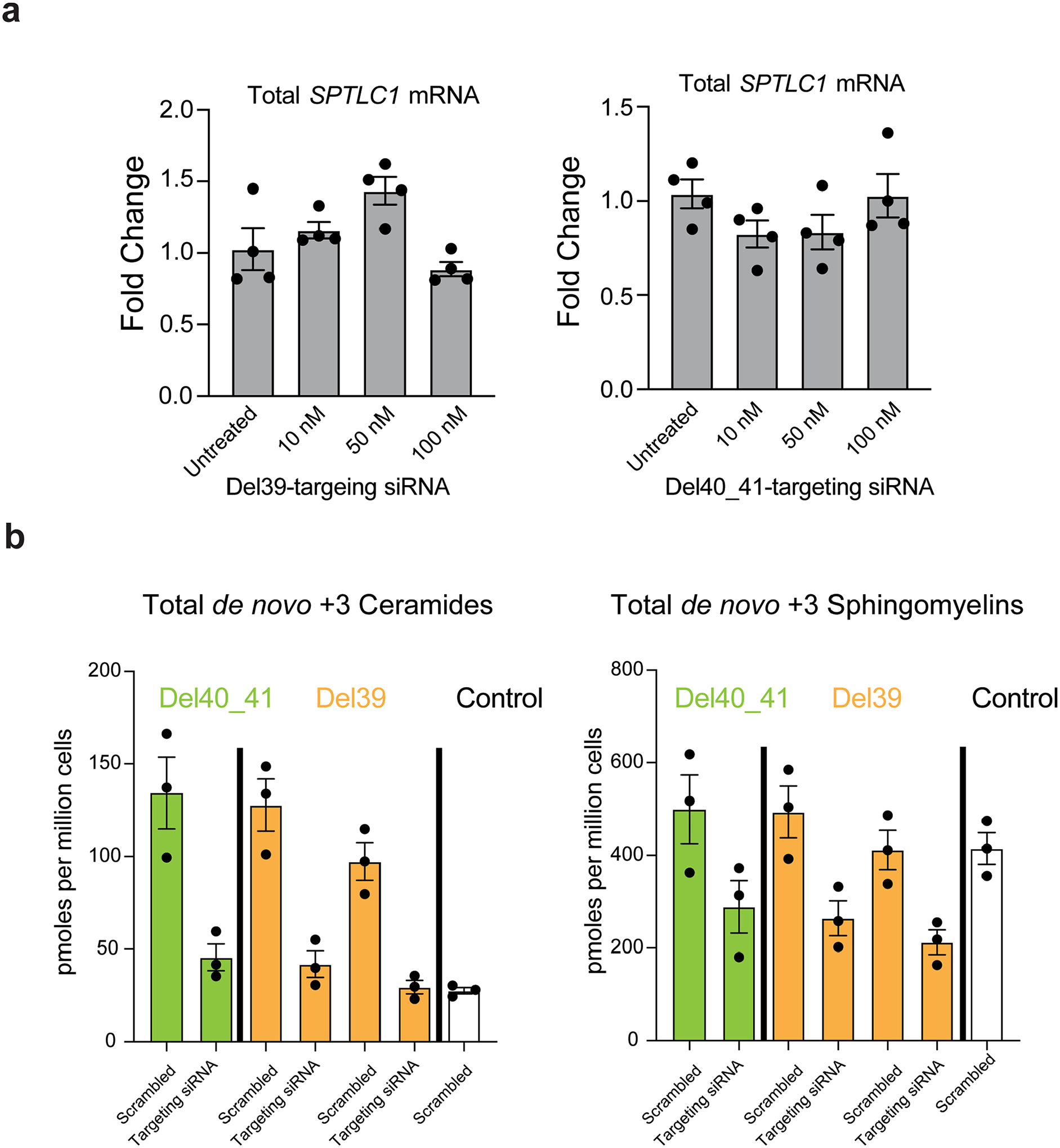
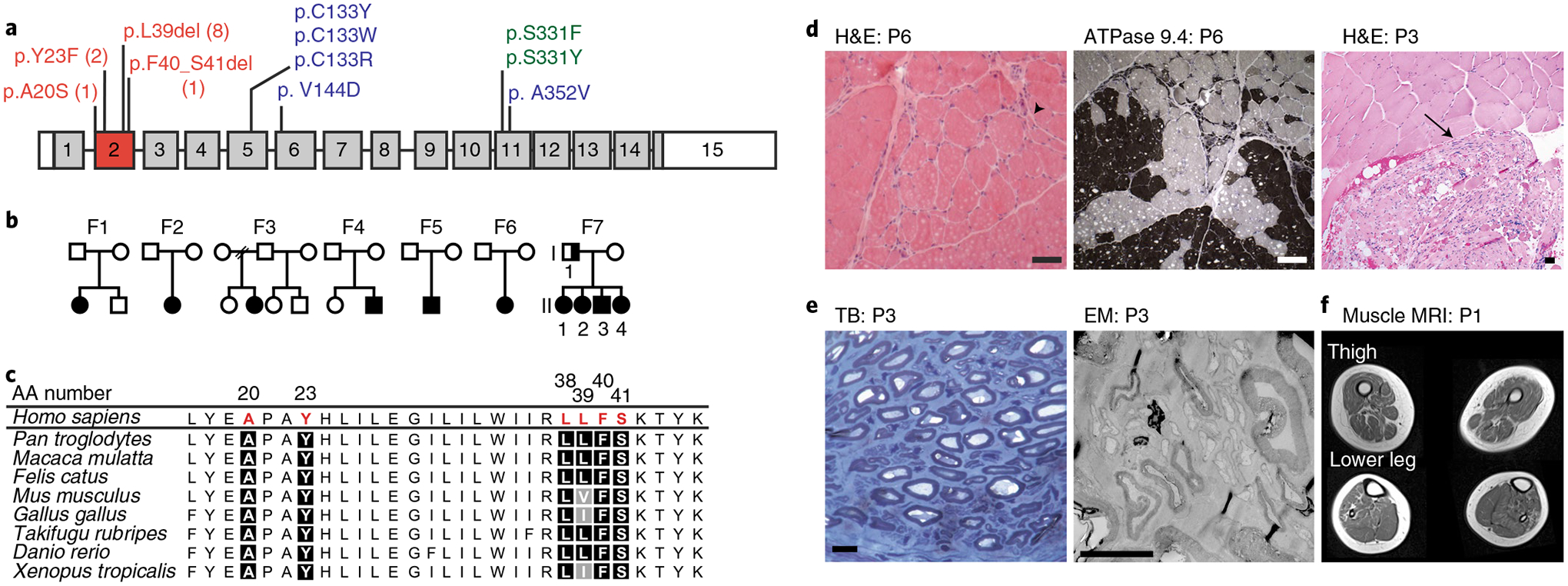
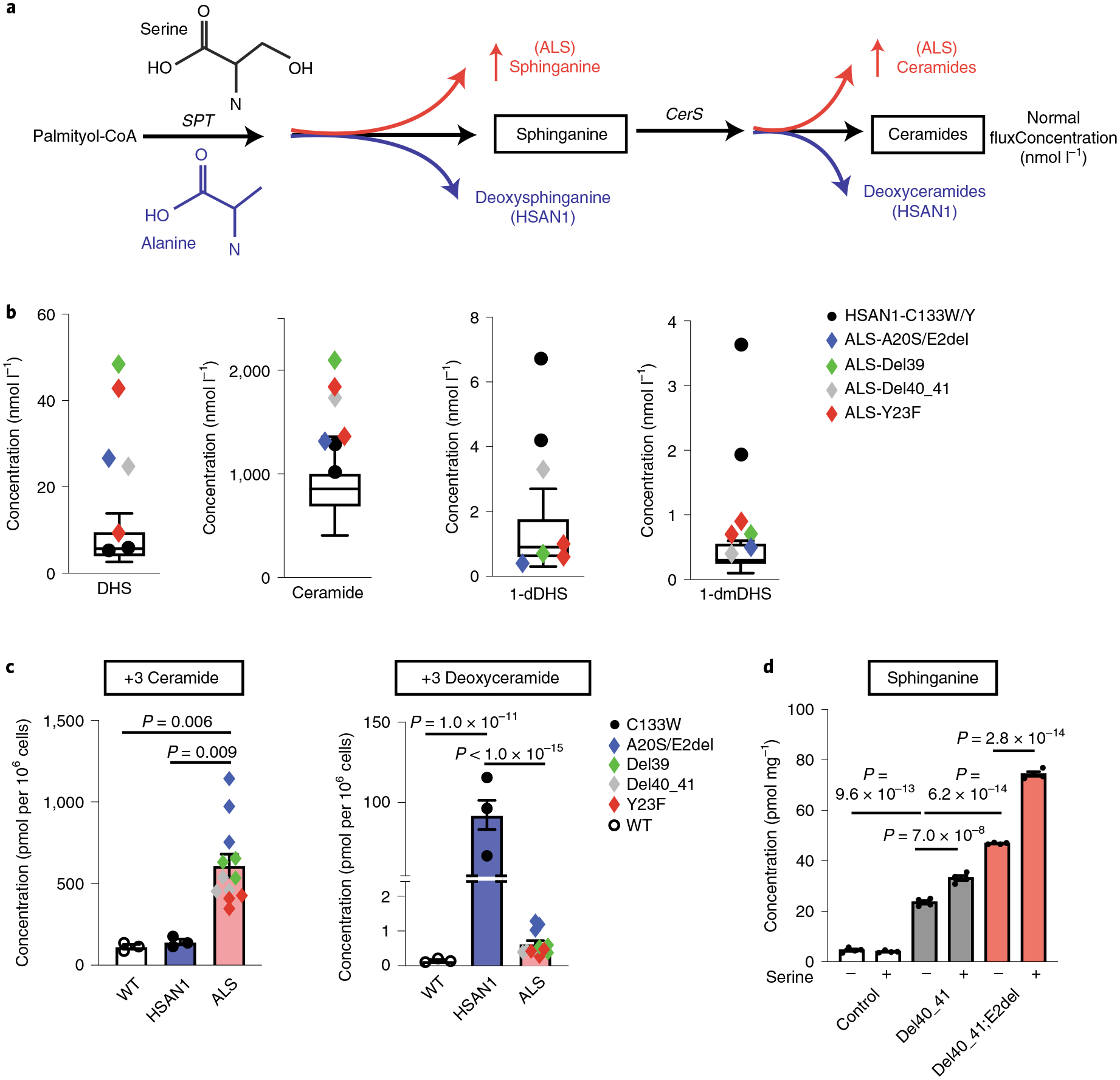
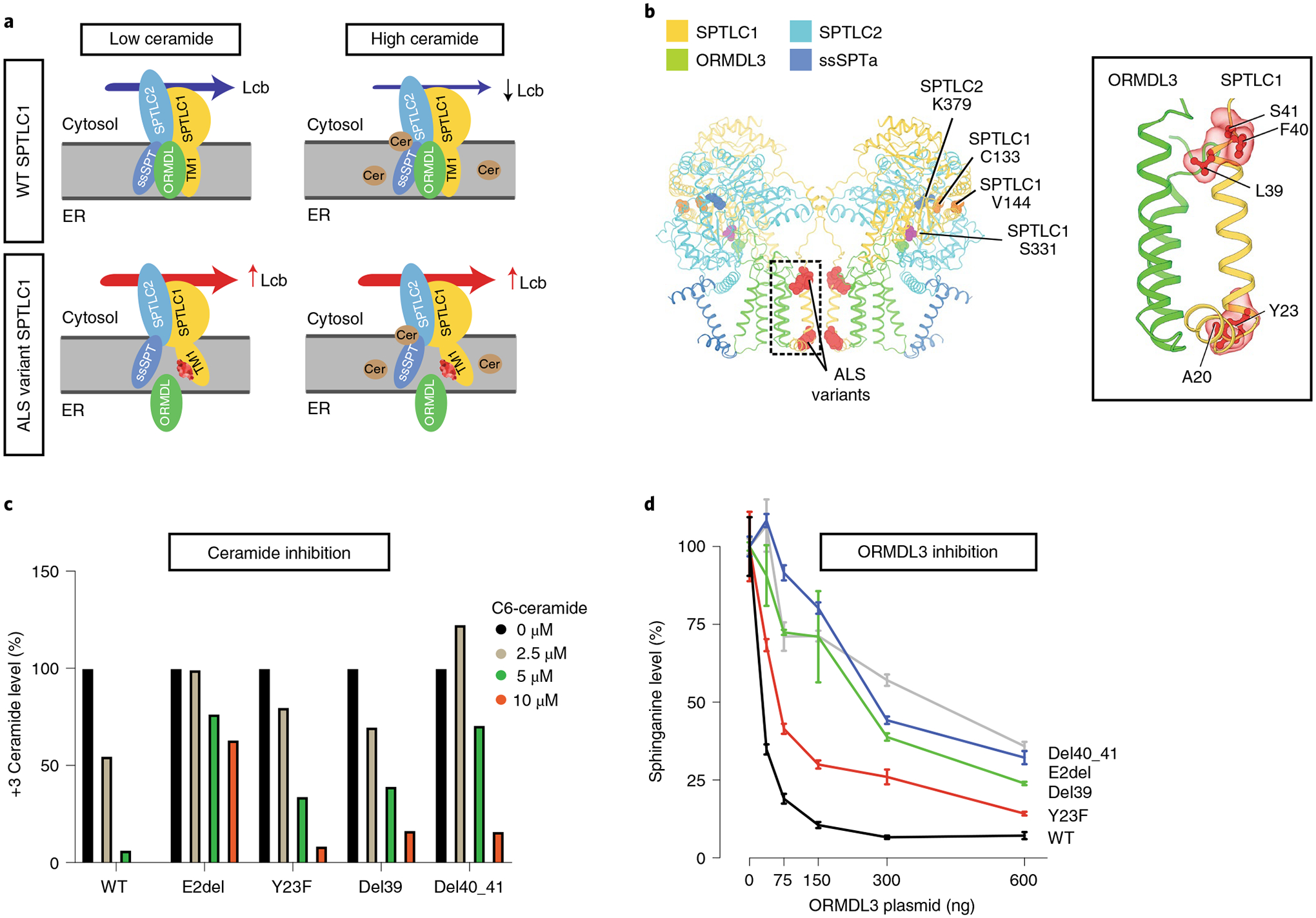
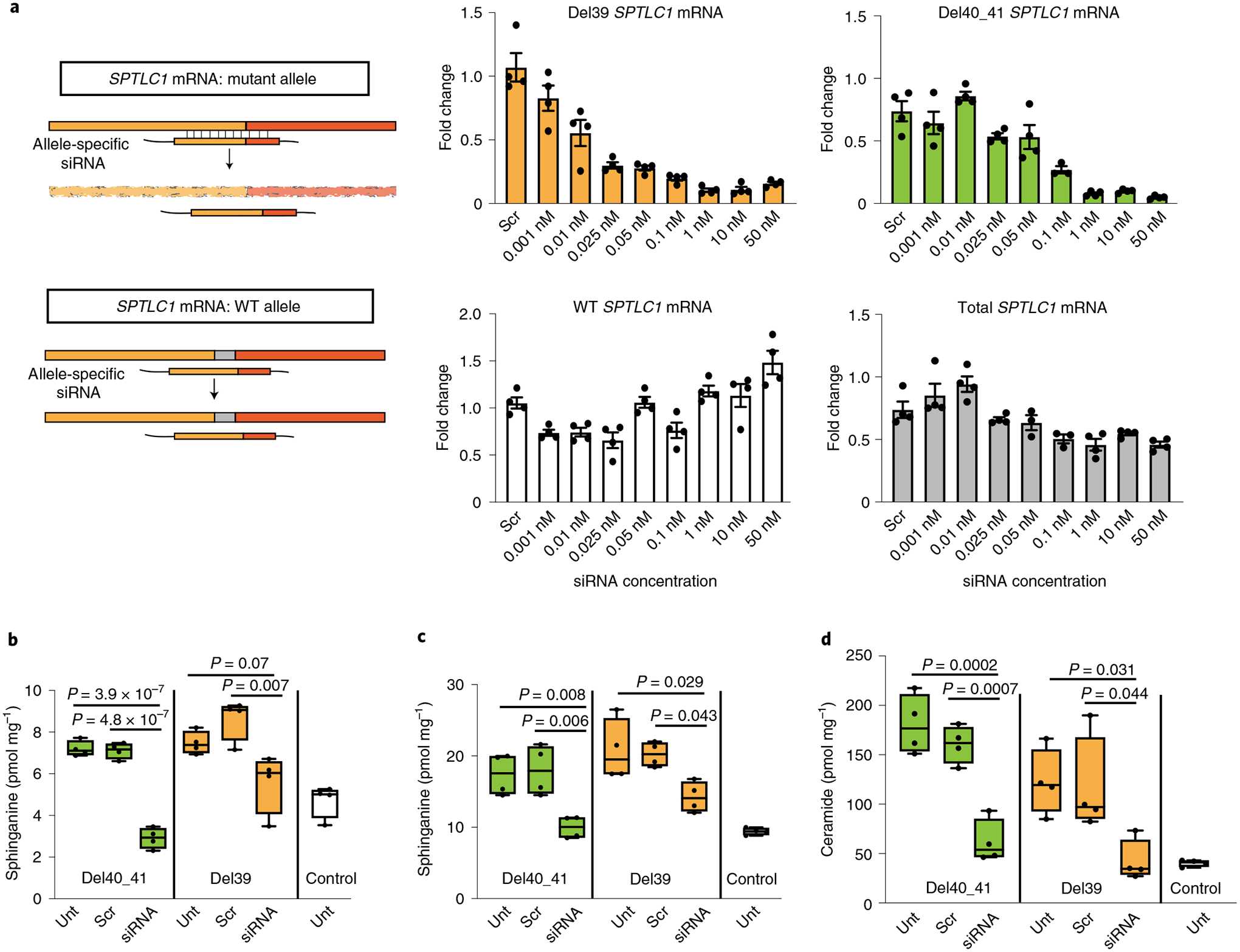
Amyotrophic lateral sclerosis (ALS) is a progressive, neurodegenerative disease of the lower and upper motor neurons with sporadic or hereditary occurrence. Age of onset, pattern of motor neuron degeneration and disease progression vary widely among individuals with ALS. Various cellular processes may drive ALS pathomechanisms, but a monogenic direct metabolic disturbance has not been causally linked to ALS. Here we show SPTLC1 variants that result in unrestrained sphingoid base synthesis cause a monogenic form of ALS. We identified four specific, dominantly acting SPTLC1 variants in seven families manifesting as childhood-onset ALS. These variants disrupt the normal homeostatic regulation of serine palmitoyltransferase (SPT) by ORMDL proteins, resulting in unregulated SPT activity and elevated levels of canonical SPT products. Notably, this is in contrast with SPTLC1 variants that shift SPT amino acid usage from serine to alanine, result in elevated levels of deoxysphingolipids and manifest with the alternate phenotype of hereditary sensory and autonomic neuropathy. We custom designed small interfering RNAs that selectively target the SPTLC1 ALS allele for degradation, leave the normal allele intact and normalize sphingolipid levels in vitro. The role of primary metabolic disturbances in ALS has been elusive; this study defines excess sphingolipid biosynthesis as a fundamental metabolic mechanism for motor neuron disease.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Comment in
-
Novel genetic form of amyotrophic lateral sclerosis reveals metabolic mechanism and therapeutic target.Nat Rev Neurol. 2021 Jul;17(7):393. doi: 10.1038/s41582-021-00528-2. Nat Rev Neurol. 2021. PMID: 34103709 No abstract available.
References
-
- Al-Chalabi A & Hardiman O The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol 9, 617–628 (2013). - PubMed
-
- Swinnen B & Robberecht W The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol 10, 661–670 (2014). - PubMed
-
- Brown RH & Al-Chalabi A Amyotrophic lateral sclerosis. N. Engl. J. Med 377, 162–172 (2017). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
