

Molecular modeling of potent novel sulfonamide derivatives as non-peptide small molecule anti-COVID 19 agents
- PMID: 34060418
- PMCID: PMC8171005
- DOI: 10.1080/07391102.2021.1897043
Molecular modeling of potent novel sulfonamide derivatives as non-peptide small molecule anti-COVID 19 agents
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent for the COVID-19. The Sulfonamides groups have been widely introduced in several drugs, especially for their antibacterial activities and generally prescribed for respiratory infections. On the other hand, imidazole groups have the multipotency to act as drugs, including antiviral activity. We have used a structure-based drug design approach to design some imidazole derivatives of sulfonamide, which can efficiently bind to the active site of SARS-CoV-2 main protease and thus may have the potential to inhibit its proteases activity. We conducted molecular docking and molecular dynamics simulation to observe the stability and flexibility of inhibitor complexes. We have checked ADMET (absorption, distribution, metabolism, excretion and toxicity) and drug-likeness rules to scrutinize toxicity and then designed the most potent compound based on computational chemistry. Our small predicted molecule non-peptide protease inhibitors could provide a useful model in the further search for novel compounds since it has many advantages over peptidic drugs, like lower side effects, toxicity and less chance of drug resistance. Further, we confirmed the stability of our inhibitor-complex and interaction profile through the Molecular dynamics simulation study. Our small predicted moleculeCommunicated by Ramaswamy H. Sarma.
Keywords: ADMET; MD simulation; SARS-CoV-2 main protease; drug-likeness; molecular docking; structure-based drug design.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
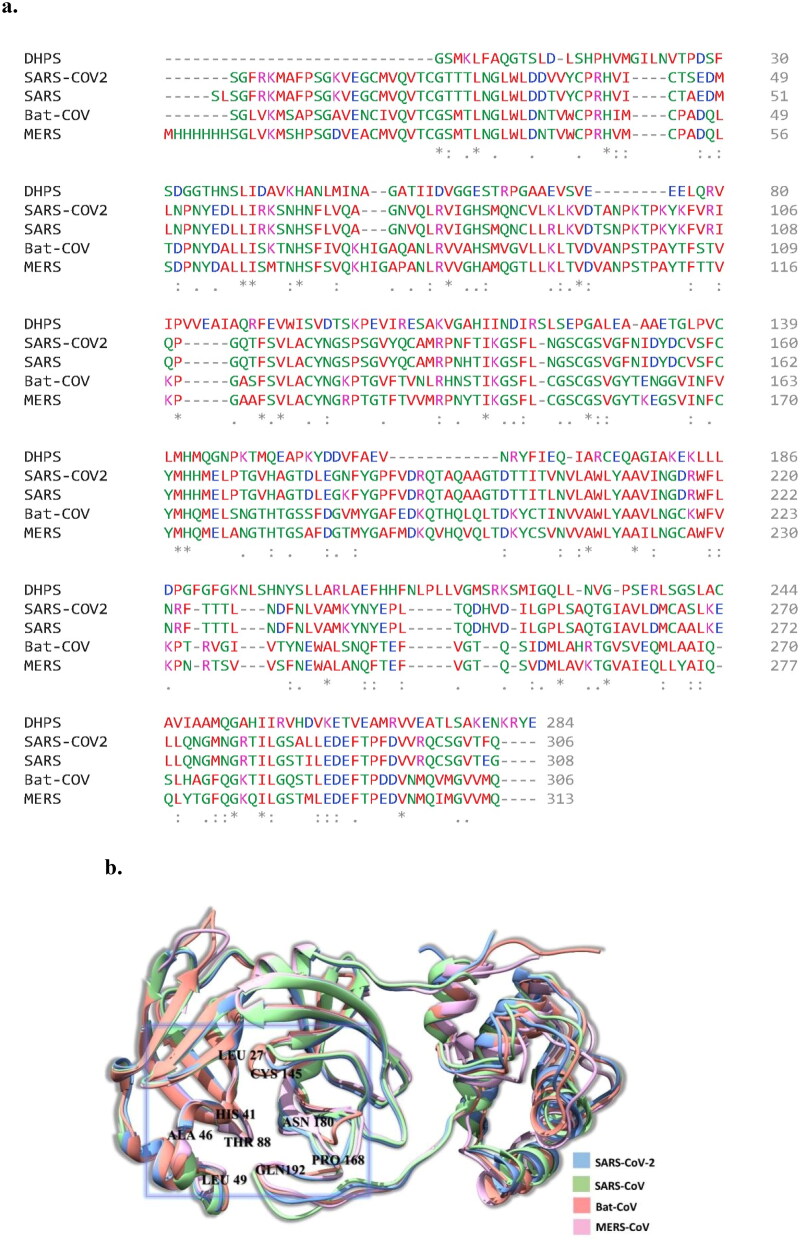
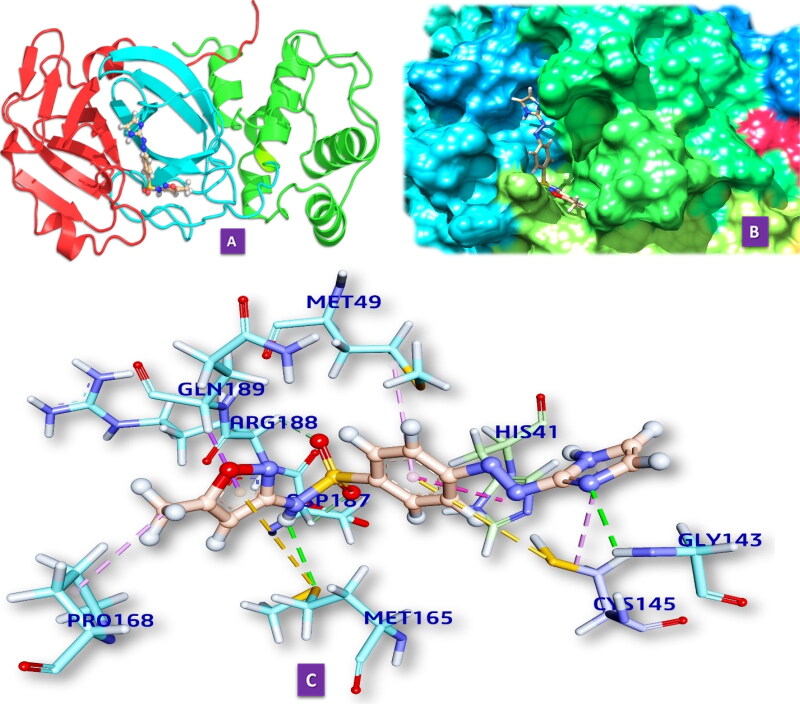
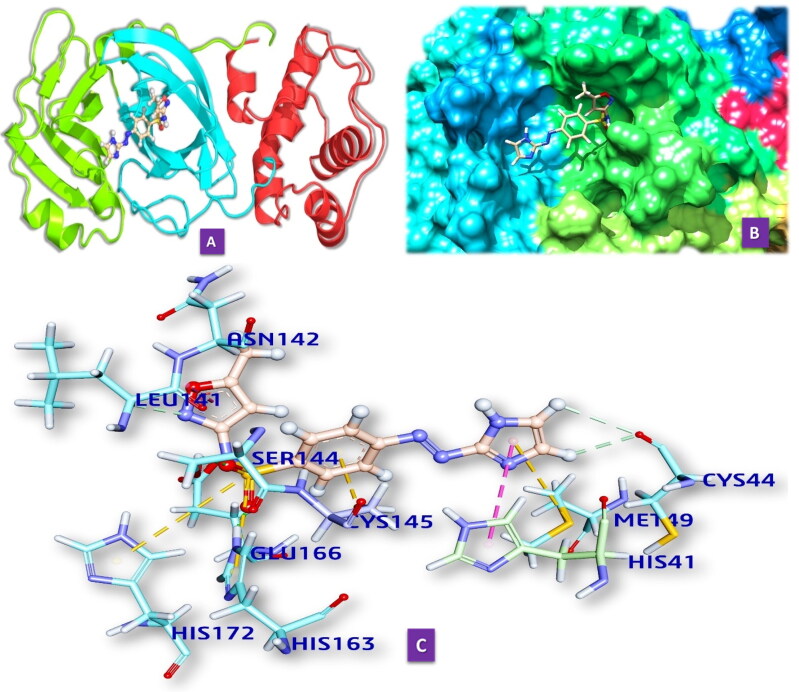
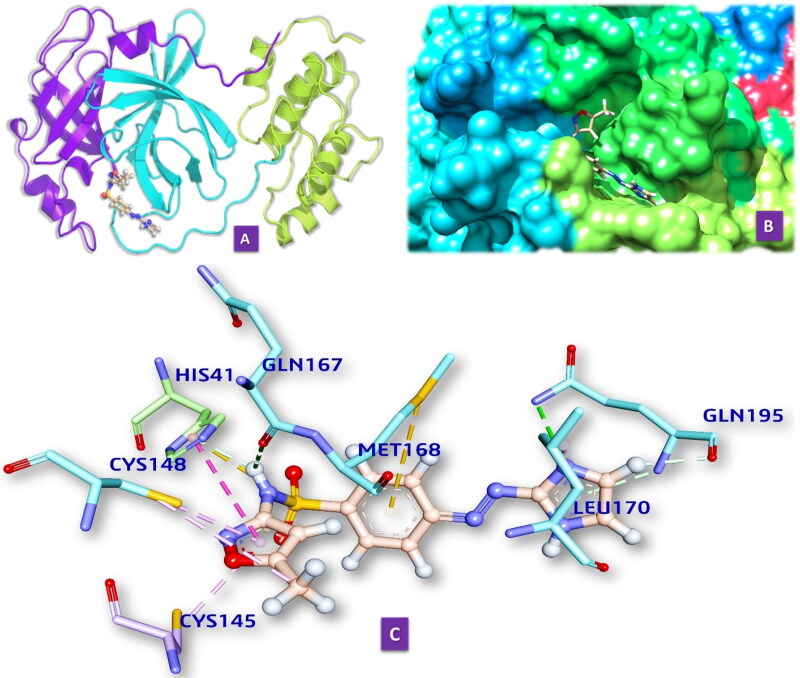
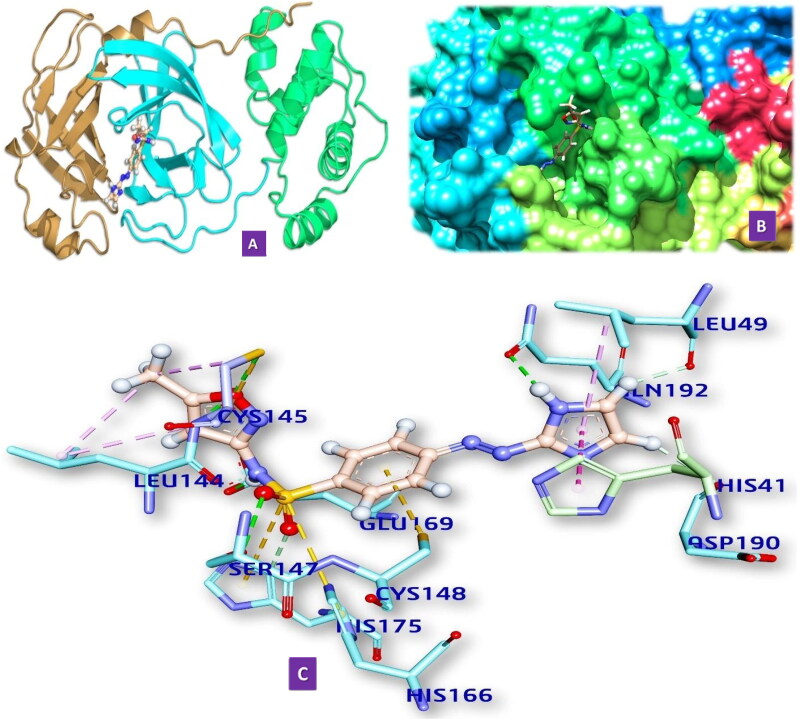
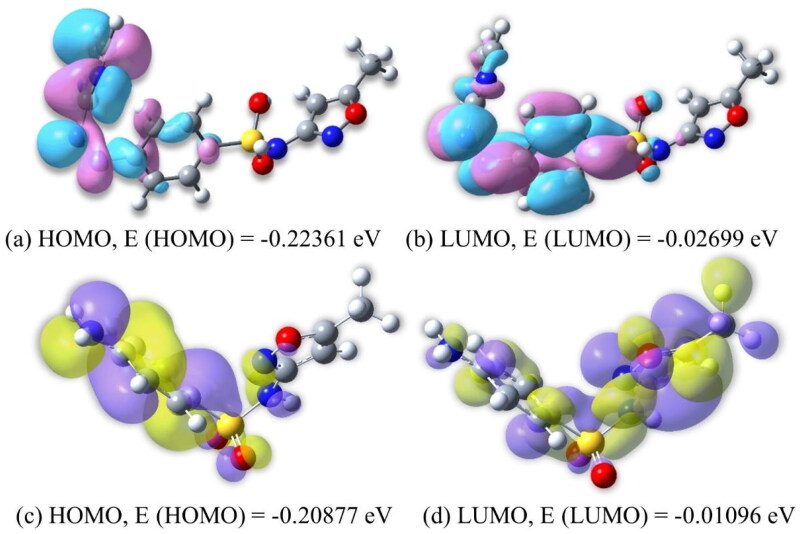
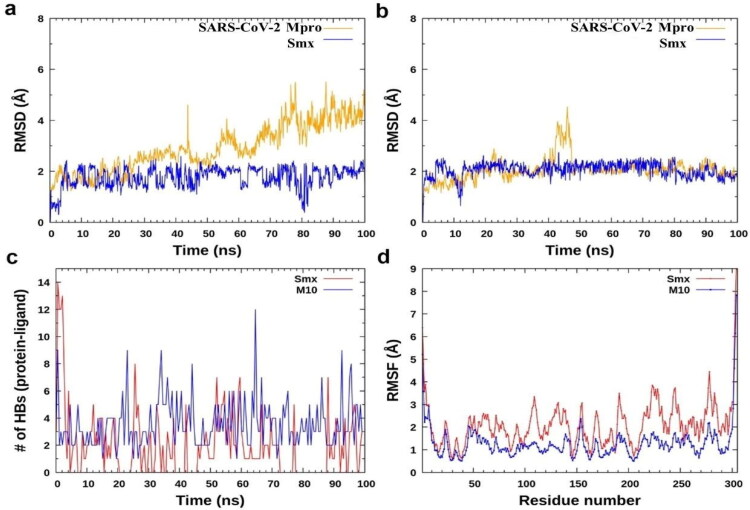
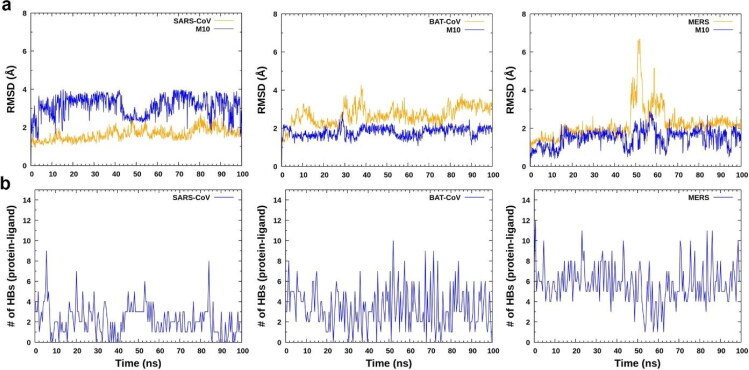
Similar articles
-
In silico analysis and identification of antiviral coumarin derivatives against 3-chymotrypsin-like main protease of the novel coronavirus SARS-CoV-2.Mol Divers. 2022 Apr;26(2):1053-1076. doi: 10.1007/s11030-021-10230-6. Epub 2021 Jul 2. Mol Divers. 2022. PMID: 34213728 Free PMC article.
-
Finding potent inhibitors against SARS-CoV-2 main protease through virtual screening, ADMET, and molecular dynamics simulation studies.J Biomol Struct Dyn. 2022 Sep;40(14):6556-6568. doi: 10.1080/07391102.2021.1897680. Epub 2021 Mar 8. J Biomol Struct Dyn. 2022. PMID: 33682642
-
Combination of QSAR, molecular docking, molecular dynamic simulation and MM-PBSA: analogues of lopinavir and favipiravir as potential drug candidates against COVID-19.J Biomol Struct Dyn. 2022 May;40(8):3711-3730. doi: 10.1080/07391102.2020.1850355. Epub 2020 Nov 30. J Biomol Struct Dyn. 2022. PMID: 33251975 Free PMC article.
-
Targeting COVID-19 (SARS-CoV-2) main protease through active phytochemicals of ayurvedic medicinal plants - Withania somnifera (Ashwagandha), Tinospora cordifolia (Giloy) and Ocimum sanctum (Tulsi) - a molecular docking study.J Biomol Struct Dyn. 2022 Jan;40(1):190-203. doi: 10.1080/07391102.2020.1810778. Epub 2020 Aug 27. J Biomol Struct Dyn. 2022. PMID: 32851919 Free PMC article.
-
Sanguiins-Promising Molecules with Broad Biological Potential.Int J Mol Sci. 2021 Nov 30;22(23):12972. doi: 10.3390/ijms222312972. Int J Mol Sci. 2021. PMID: 34884795 Free PMC article. Review.
References
-
- Aihara, J.-I. (2000). Correlation found between the HOMO–LUMO energy separation and the chemical reactivity at the most reactive site for isolated-pentagon isomers of fullerenes. Physical Chemistry Chemical Physics, 2(14), 3121–3125. 10.1039/b002601h - DOI
-
- Ajay Bemis, G. W., & Murcko, M. A. (1999). Designing libraries with CNS activity. Journal of Medicinal Chemistry, 42(24), 4942–4951. - PubMed
-
- Bano, S., Javed, K., Ahmad, S., Rathish, I., Singh, S., & Alam, M. (2011). Synthesis and biological evaluation of some new 2-pyrazolines bearing benzene sulfonamide moiety as potential anti-inflammatory and anti-cancer agents. European Journal of Medicinal Chemistry, 46(12), 5763–5768. 10.1016/j.ejmech.2011.08.015 - DOI - PubMed
-
- Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. A., & Karplus, M. (1983). CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, 4(2), 187–217. 10.1002/jcc.540040211 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous