

GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection
- PMID: 34061598
- PMCID: PMC8262878
- DOI: 10.1128/mBio.03269-20
GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection
Abstract
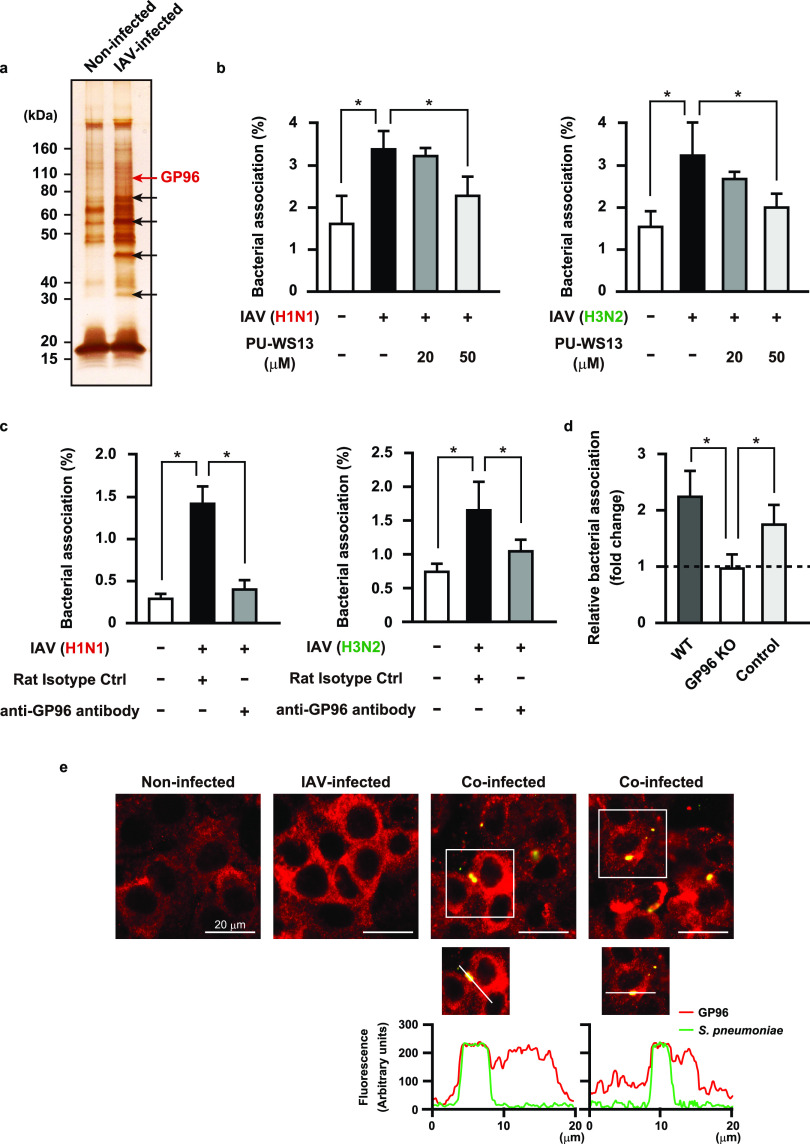
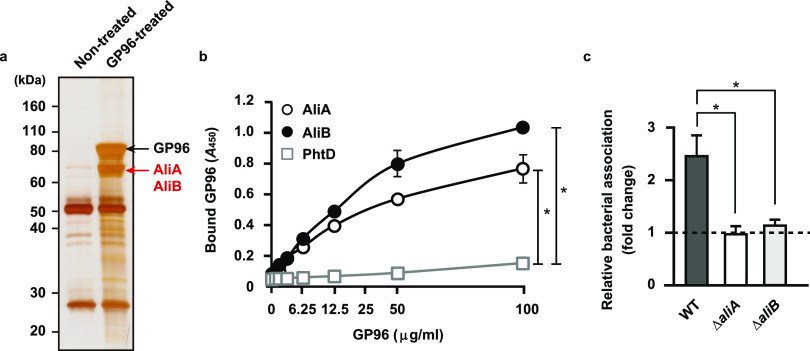
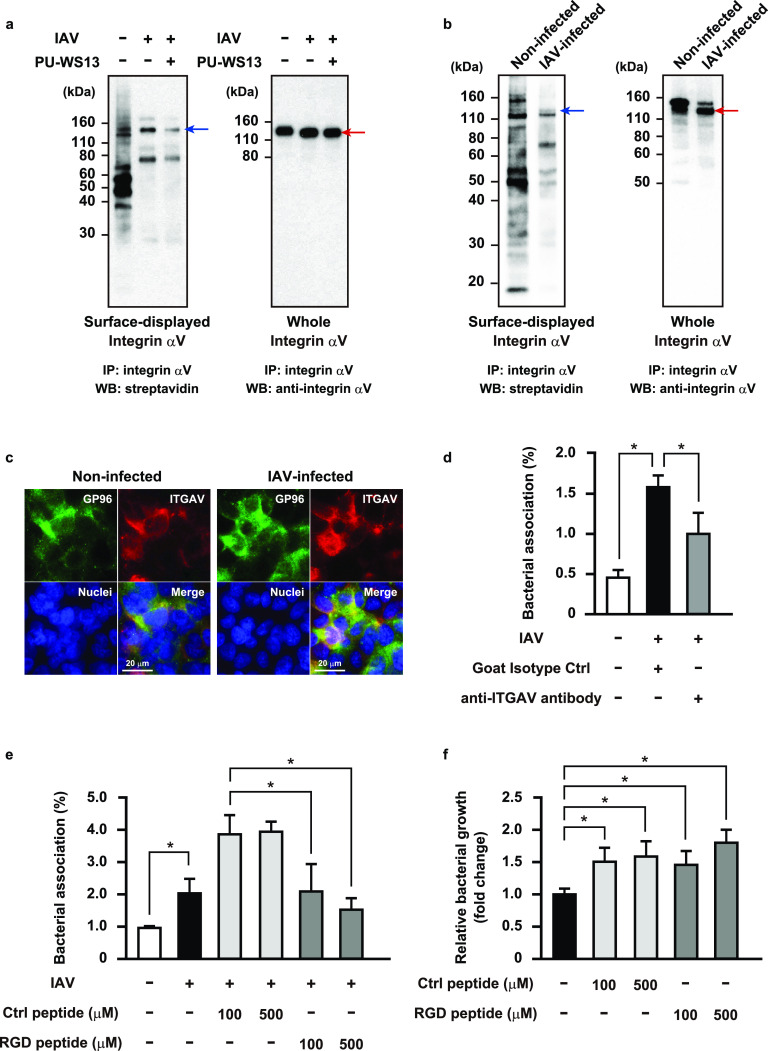
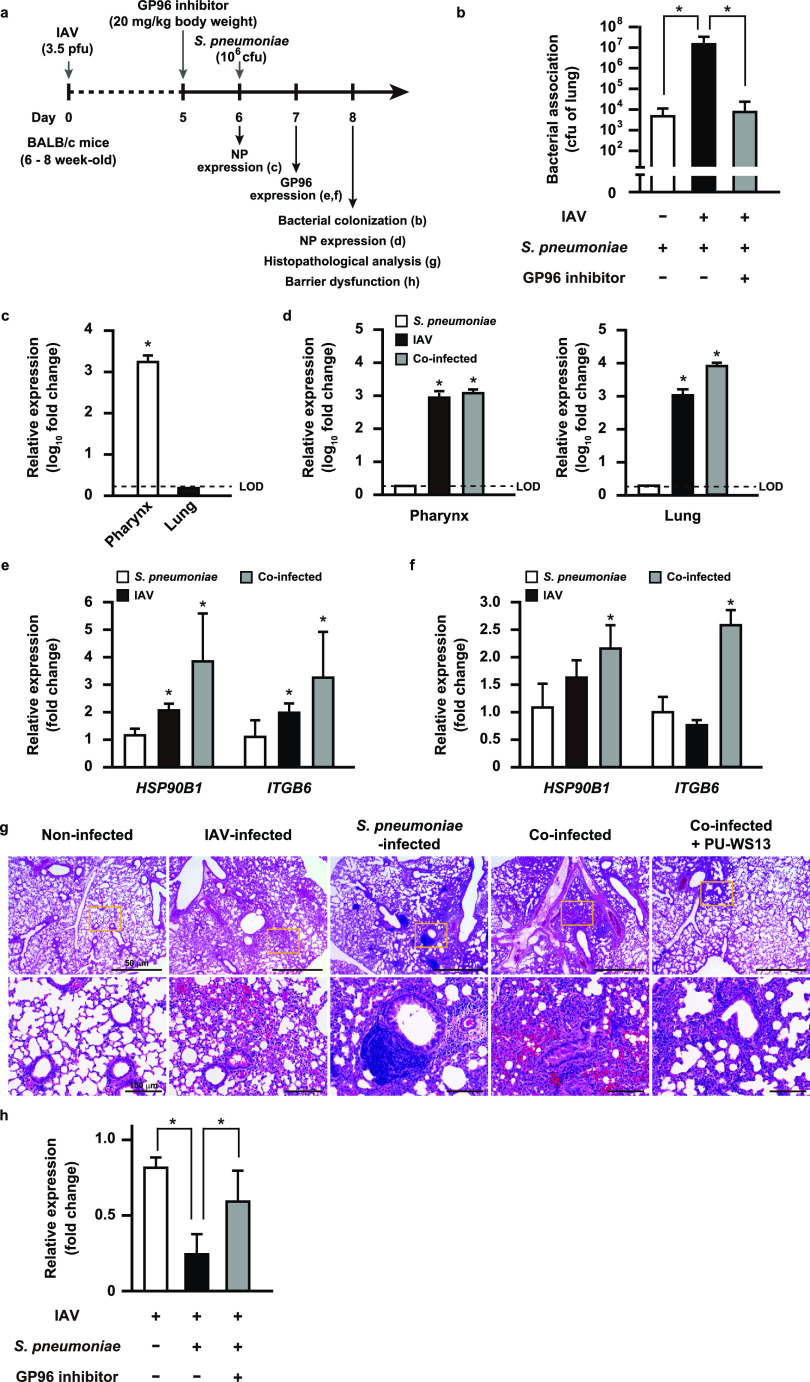
Influenza A virus (IAV) infection predisposes the host to secondary bacterial pneumonia, known as a major cause of morbidity and mortality during influenza virus epidemics. Analysis of interactions between IAV-infected human epithelial cells and Streptococcus pneumoniae revealed that infected cells ectopically exhibited the endoplasmic reticulum chaperone glycoprotein 96 (GP96) on the surface. Importantly, efficient pneumococcal adherence to epithelial cells was imparted by interactions with extracellular GP96 and integrin αV, with the surface expression mediated by GP96 chaperone activity. Furthermore, abrogation of adherence was gained by chemical inhibition or genetic knockout of GP96 as well as addition of RGD peptide, an inhibitor of integrin-ligand interactions. Direct binding of extracellular GP96 and pneumococci was shown to be mediated by pneumococcal oligopeptide permease components. Additionally, IAV infection induced activation of calpains and Snail1, which are responsible for degradation and transcriptional repression of junctional proteins in the host, respectively, indicating increased bacterial translocation across the epithelial barrier. Notably, treatment of IAV-infected mice with the GP96 inhibitor enhanced pneumococcal clearance from lung tissues and ameliorated lung pathology. Taken together, the present findings indicate a viral-bacterial synergy in relation to disease progression and suggest a paradigm for developing novel therapeutic strategies tailored to inhibit pneumococcal colonization in an IAV-infected respiratory tract. IMPORTANCE Secondary bacterial pneumonia following an influenza A virus (IAV) infection is a major cause of morbidity and mortality. Although it is generally accepted that preceding IAV infection leads to increased susceptibility to secondary bacterial infection, details regarding the pathogenic mechanism during the early stage of superinfection remain elusive. Here, we focused on the interaction of IAV-infected cells and Streptococcus pneumoniae, which revealed that human epithelial cells infected with IAV exhibit a cell surface display of GP96, an endoplasmic reticulum chaperon. Notably, extracellular GP96 was shown to impart efficient adherence for secondary infection by S. pneumoniae, and GP96 inhibition ameliorated lung pathology of superinfected mice, indicating it to be a useful target for development of therapeutic strategies for patients with superinfection.
Keywords: Streptococcus pneumoniae; influenza virus; pneumonia; superinfection.
Figures

Similar articles
-
Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors.Proc Natl Acad Sci U S A. 2015 Jan 6;112(1):238-43. doi: 10.1073/pnas.1414422112. Epub 2014 Dec 22. Proc Natl Acad Sci U S A. 2015. PMID: 25535343 Free PMC article.
-
Cysteinyl Maresins Reprogram Macrophages to Protect Mice from Streptococcus pneumoniae after Influenza A Virus Infection.mBio. 2022 Aug 30;13(4):e0126722. doi: 10.1128/mbio.01267-22. Epub 2022 Aug 1. mBio. 2022. PMID: 35913160 Free PMC article.
-
Memory Th17 cell-mediated protection against lethal secondary pneumococcal pneumonia following influenza infection.mBio. 2023 Aug 31;14(4):e0051923. doi: 10.1128/mbio.00519-23. Epub 2023 May 24. mBio. 2023. PMID: 37222516 Free PMC article.
-
Secondary streptococcal infection following influenza.Microbiol Immunol. 2022 Jun;66(6):253-263. doi: 10.1111/1348-0421.12965. Epub 2022 May 26. Microbiol Immunol. 2022. PMID: 35088451 Review.
-
Interactions between Streptococcus pneumoniae and influenza virus: a mutually beneficial relationship?Future Microbiol. 2012 May;7(5):609-24. doi: 10.2217/fmb.12.29. Future Microbiol. 2012. PMID: 22568716 Review.
Cited by
-
Effect of glucocorticoid combined with azithromycin on serum inflammatory factors and pulmonary function in children with influenza A virus-induced pneumonia.Medicine (Baltimore). 2025 Jun 6;104(23):e42117. doi: 10.1097/MD.0000000000042117. Medicine (Baltimore). 2025. PMID: 40489830 Free PMC article.
-
Targeting stressor-induced dysfunctions in protein-protein interaction networks via epichaperomes.Trends Pharmacol Sci. 2023 Jan;44(1):20-33. doi: 10.1016/j.tips.2022.10.006. Epub 2022 Nov 20. Trends Pharmacol Sci. 2023. PMID: 36414432 Free PMC article. Review.
-
Unraveling the Mechanism of Epichaperome Modulation by Zelavespib: Biochemical Insights on Target Occupancy and Extended Residence Time at the Site of Action.Biomedicines. 2023 Sep 22;11(10):2599. doi: 10.3390/biomedicines11102599. Biomedicines. 2023. PMID: 37892973 Free PMC article.
-
The Role of TLR4 in Lung Epithelial Cell Injury Caused by Influenza Virus Combined with Staphylococcus aureus.Microorganisms. 2025 May 24;13(6):1201. doi: 10.3390/microorganisms13061201. Microorganisms. 2025. PMID: 40572089 Free PMC article.
-
Pneumolysin contributes to dysfunction of nasal epithelial barrier for promotion of pneumococcal dissemination into brain tissue.mSphere. 2024 Oct 29;9(10):e0065524. doi: 10.1128/msphere.00655-24. Epub 2024 Sep 30. mSphere. 2024. PMID: 39345124 Free PMC article.
References
-
- Zhang Q, Shi J, Deng G, Guo J, Zeng X, He X, Kong H, Gu C, Li X, Liu J, Wang G, Chen Y, Liu L, Liang L, Li Y, Fan J, Wang J, Li W, Guan L, Li Q, Yang H, Chen P, Jiang L, Guan Y, Xin X, Jiang Y, Tian G, Wang X, Qiao C, Li C, Bu Z, Chen H. 2013. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 341:410–414. doi:10.1126/science.1240532. - DOI - PubMed
-
- Liang L, Jiang L, Li J, Zhao Q, Wang J, He X, Huang S, Wang Q, Zhao Y, Wang G, Sun N, Deng G, Shi J, Tian G, Zeng X, Jiang Y, Liu L, Liu J, Chen P, Bu Z, Kawaoka Y, Chen H, Li C. 2019. Low polymerase activity attributed to PA drives the acquisition of the PB2 E627K mutation of H7N9 avian influenza virus in mammals. mBio 10:e01162-19. doi:10.1128/mBio.01162-19. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
