

Integrated analysis of multimodal single-cell data
- PMID: 34062119
- PMCID: PMC8238499
- DOI: 10.1016/j.cell.2021.04.048
Integrated analysis of multimodal single-cell data
Abstract
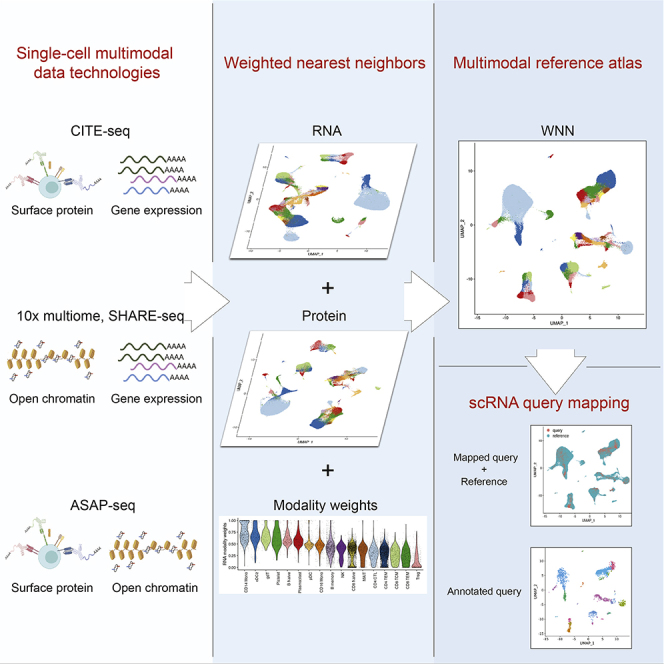
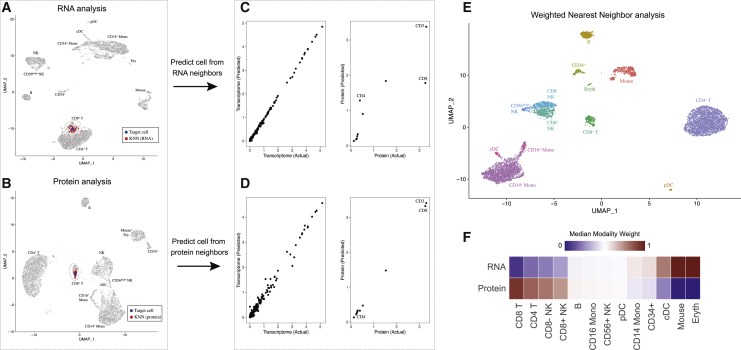
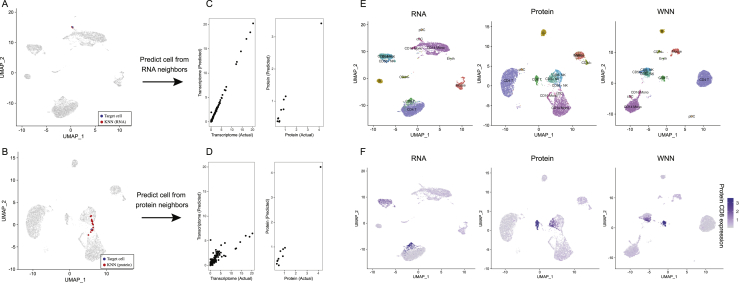
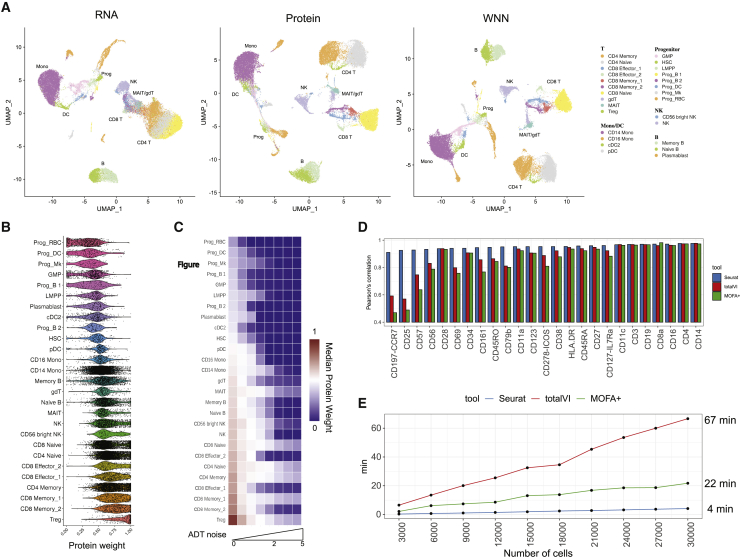
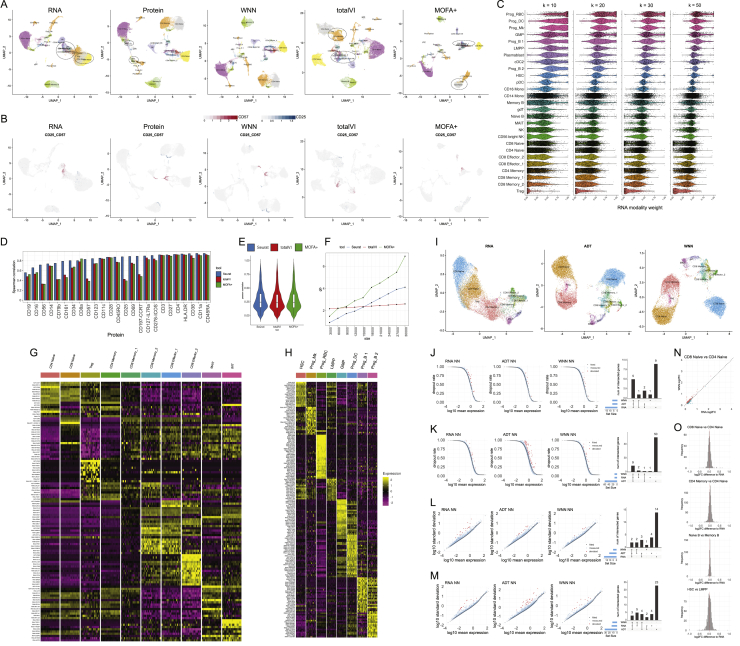
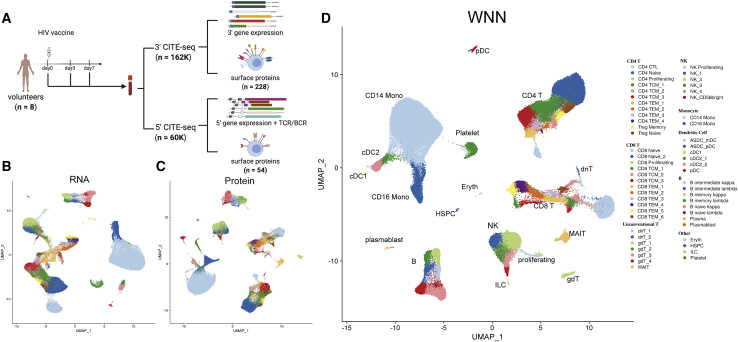
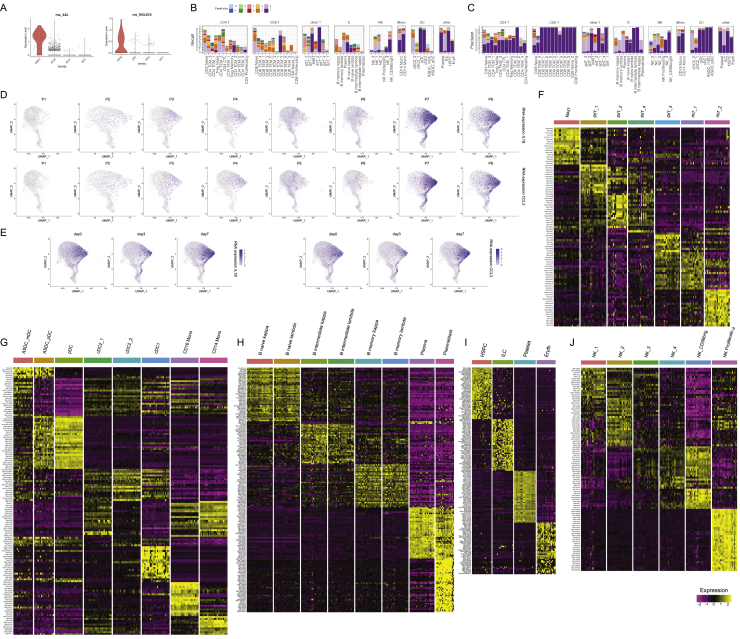
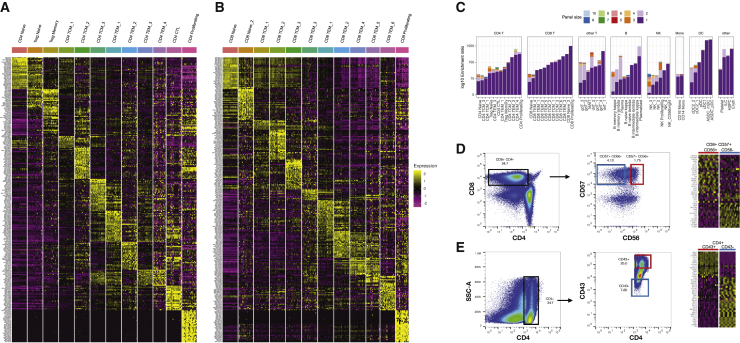
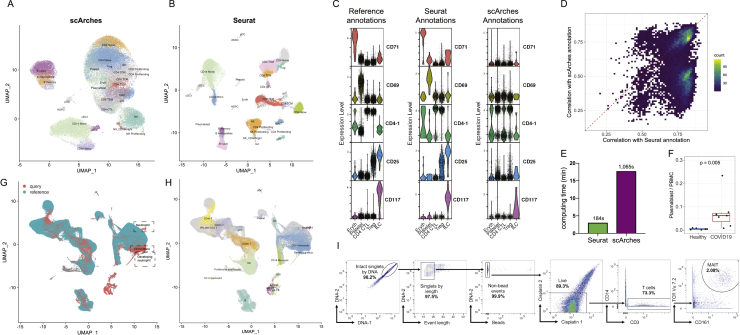
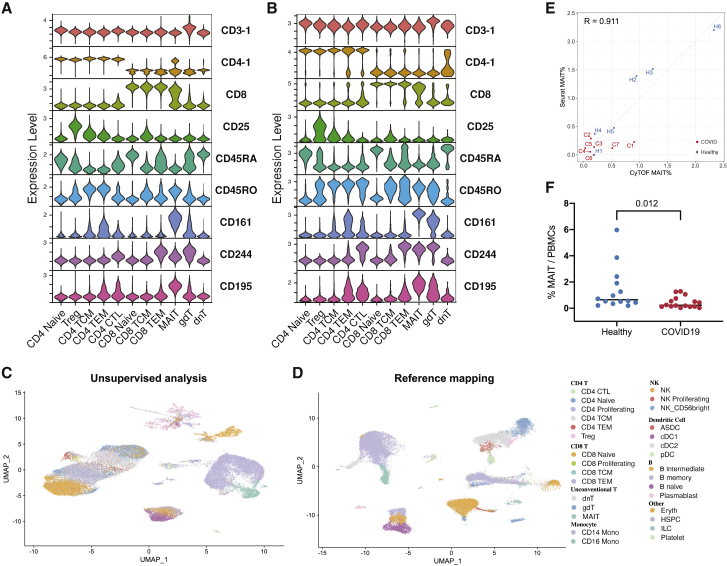
The simultaneous measurement of multiple modalities represents an exciting frontier for single-cell genomics and necessitates computational methods that can define cellular states based on multimodal data. Here, we introduce "weighted-nearest neighbor" analysis, an unsupervised framework to learn the relative utility of each data type in each cell, enabling an integrative analysis of multiple modalities. We apply our procedure to a CITE-seq dataset of 211,000 human peripheral blood mononuclear cells (PBMCs) with panels extending to 228 antibodies to construct a multimodal reference atlas of the circulating immune system. Multimodal analysis substantially improves our ability to resolve cell states, allowing us to identify and validate previously unreported lymphoid subpopulations. Moreover, we demonstrate how to leverage this reference to rapidly map new datasets and to interpret immune responses to vaccination and coronavirus disease 2019 (COVID-19). Our approach represents a broadly applicable strategy to analyze single-cell multimodal datasets and to look beyond the transcriptome toward a unified and multimodal definition of cellular identity.
Keywords: CITE-seq; COVID-19; T cell; immune system; multimodal analysis; reference mapping; single cell genomics.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests In the past three years, R.S. has worked as a consultant for Bristol-Myers Squibb, Regeneron, and Kallyope and served as an SAB member for ImmunAI, Apollo Life Sciences GmbH, Nanostring, and the NYC Pandemic Response Lab. R.G. has received consulting income from Juno Therapeutics, Takeda, Infotech Soft, Celgene, and Merck, he has received research support from Janssen Pharmaceuticals and Juno Therapeutics, and he declares ownership in CellSpace Biosciences. P.S. is a co-inventor of a patent related to this work. B.Z.Y. is an employee at BioLegend Inc., which is the exclusive licensee of the New York Genome Center patent application related to this work.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
