

Molecular, Cellular and Functional Changes in the Retinas of Young Adult Mice Lacking the Voltage-Gated K+ Channel Subunits Kv8.2 and K2.1
- PMID: 34063002
- PMCID: PMC8124447
- DOI: 10.3390/ijms22094877
Molecular, Cellular and Functional Changes in the Retinas of Young Adult Mice Lacking the Voltage-Gated K+ Channel Subunits Kv8.2 and K2.1
Abstract
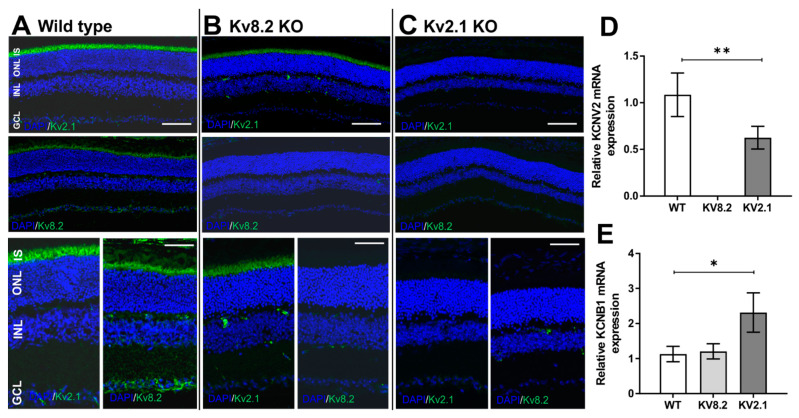
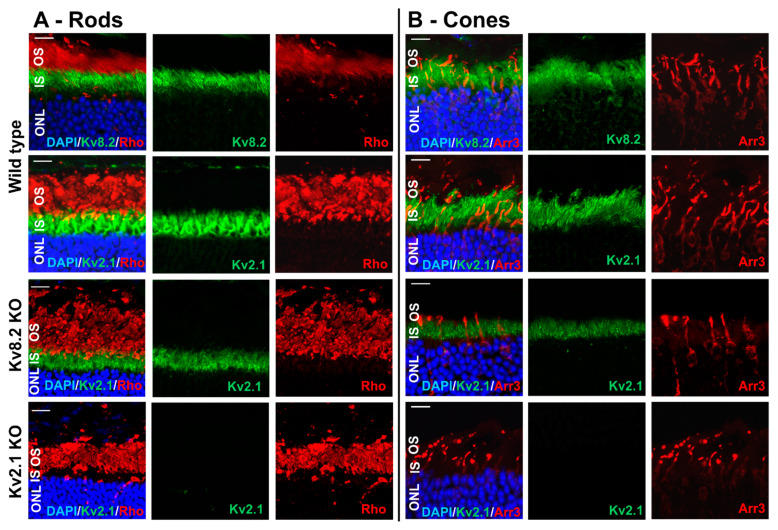
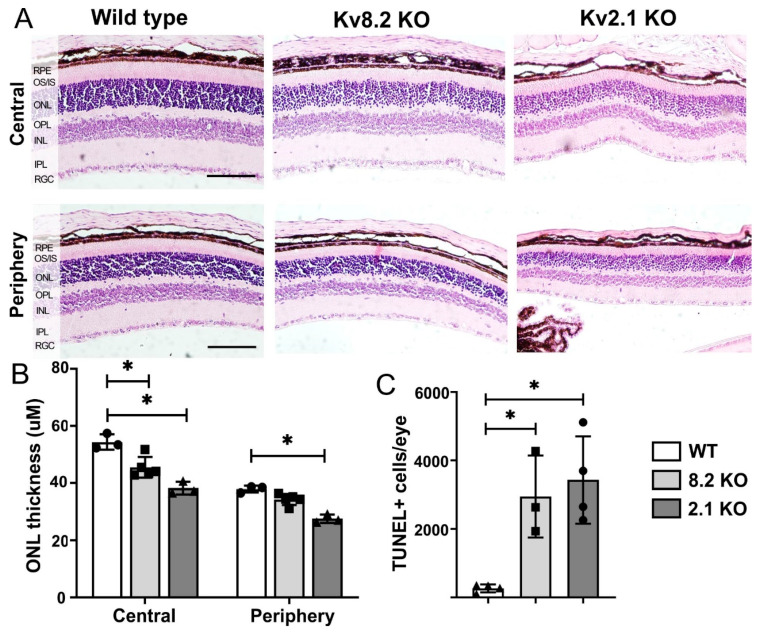
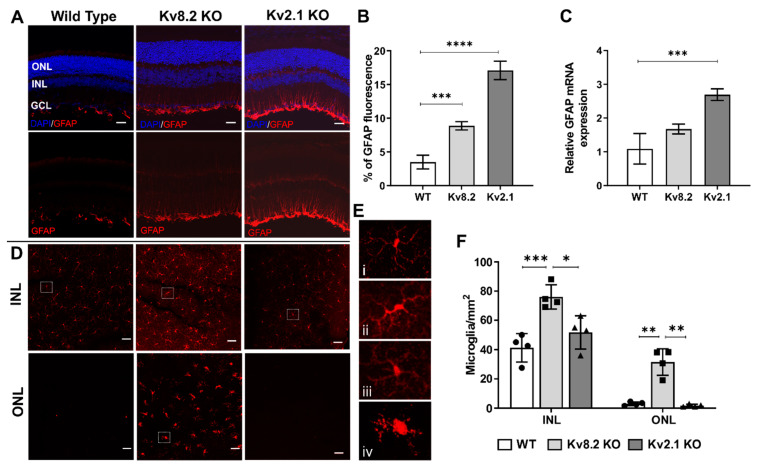
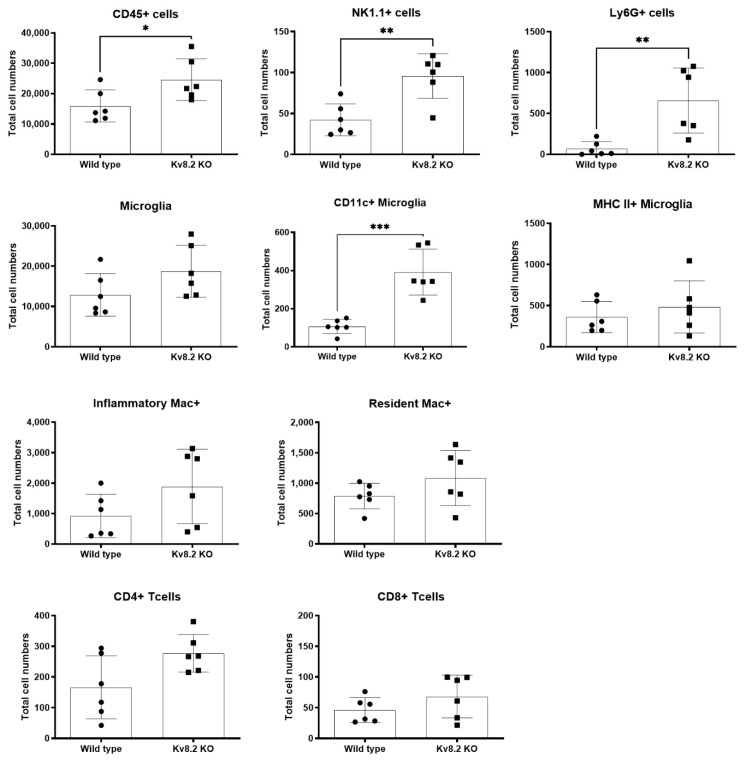
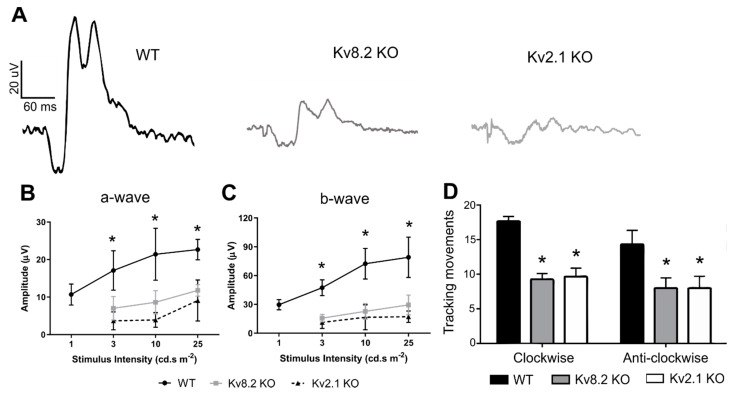
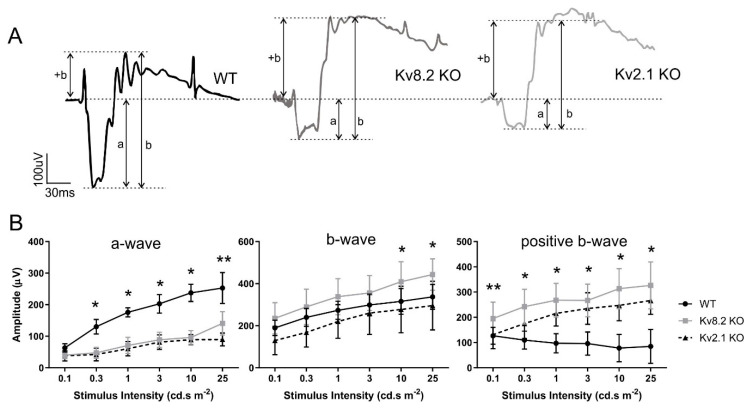
Cone Dystrophy with Supernormal Rod Response (CDSRR) is a rare autosomal recessive disorder leading to severe visual impairment in humans, but little is known about its unique pathophysiology. We have previously shown that CDSRR is caused by mutations in the KCNV2 (Potassium Voltage-Gated Channel Modifier Subfamily V Member 2) gene encoding the Kv8.2 subunit, a modulatory subunit of voltage-gated potassium (Kv) channels. In a recent study, we validated a novel mouse model of Kv8.2 deficiency at a late stage of the disease and showed that it replicates the human electroretinogram (ERG) phenotype. In this current study, we focused our investigation on young adult retinas to look for early markers of disease and evaluate their effect on retinal morphology, electrophysiology and immune response in both the Kv8.2 knockout (KO) mouse and in the Kv2.1 KO mouse, the obligate partner of Kv8.2 in functional retinal Kv channels. By evaluating the severity of retinal dystrophy in these KO models, we demonstrated that retinas of Kv KO mice have significantly higher apoptotic cells, a thinner outer nuclear cell layer and increased activated microglia cells in the subretinal space. Our results indicate that in the murine retina, the loss of Kv8.2 subunits contributes to early cellular and physiological changes leading to retinal dysfunction. These results could have potential implications in the early management of CDSRR despite its relatively nonprogressive nature in humans.
Keywords: CDSRR; KCNB1; KCNV2; cone-rod dystrophy; photoreceptors; retinal degeneration; voltage-gated potassium channels.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Wu H., Cowing J.A., Michaelides M., Wilkie S.E., Jeffery G., Jenkins S.A., Mester V., Bird A.C., Robson A.G., Holder G.E., et al. Mutations in the Gene KCNV2 Encoding a Voltage-Gated Potassium Channel Subunit Cause “Cone Dystrophy with Supernormal Rod Electroretinogram” in Humans. Am. J. Hum. Genet. 2006;79:574–579. doi: 10.1086/507568. - DOI - PMC - PubMed
-
- Wissinger B., Dangel S., Jagle H., Hansen L., Baumann B., Rudolph G., Wolf C., Bonin M., Koeppen K., Ladewig T., et al. Cone Dystrophy with Supernormal Rod Response Is Strictly Associated with Mutations in KCNV2. Investig. Opthalmol. Vis. Sci. 2008;49:751–757. doi: 10.1167/iovs.07-0471. - DOI - PubMed
-
- Wissinger B., Schaich S., Baumann B., Bonin M., Jägle H., Friedburg C., Varsányi B., Hoyng C.B., Dollfus H., Heckenlively J.R., et al. Large deletions of the KCNV2 gene are common in patients with cone dystrophy with supernormal rod response. Hum. Mutat. 2011;32:1398–1406. doi: 10.1002/humu.21580. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
