

Enhanced Efficacy of Aurora Kinase Inhibitors in G2/M Checkpoint Deficient TP53 Mutant Uterine Carcinomas Is Linked to the Summation of LKB1-AKT-p53 Interactions
- PMID: 34063609
- PMCID: PMC8125555
- DOI: 10.3390/cancers13092195
Enhanced Efficacy of Aurora Kinase Inhibitors in G2/M Checkpoint Deficient TP53 Mutant Uterine Carcinomas Is Linked to the Summation of LKB1-AKT-p53 Interactions
Abstract
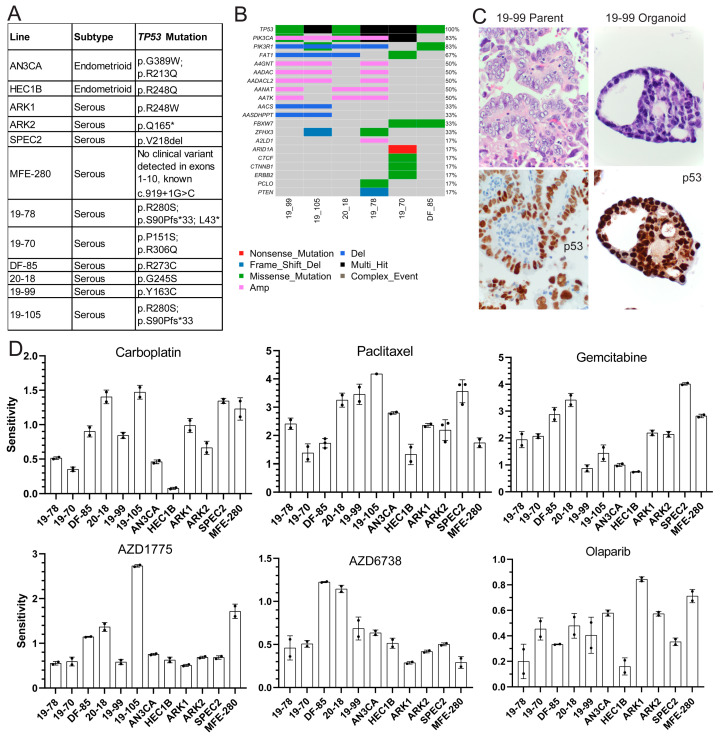
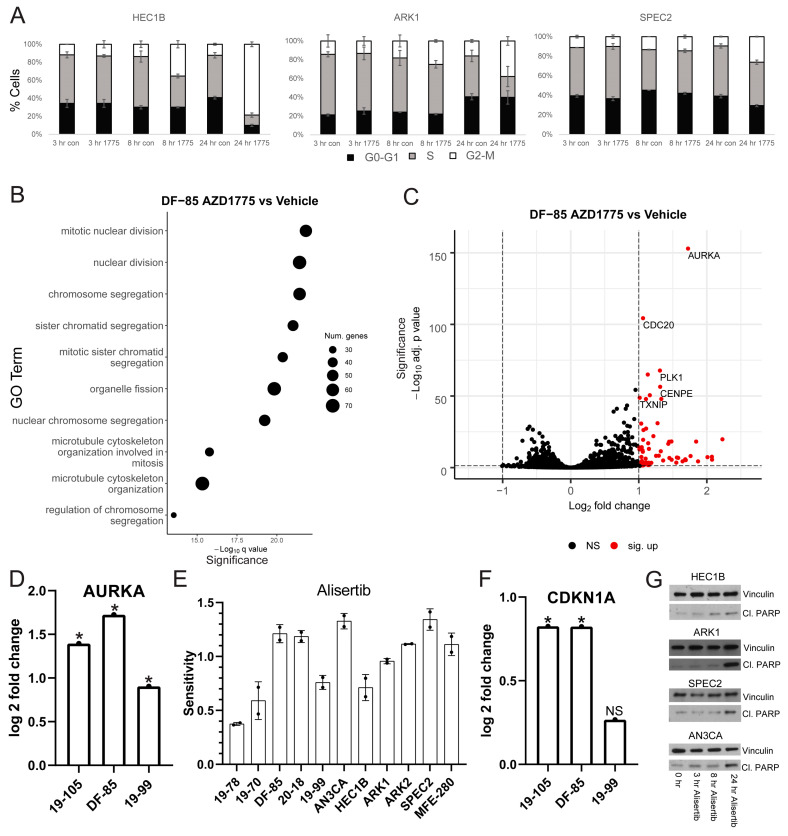
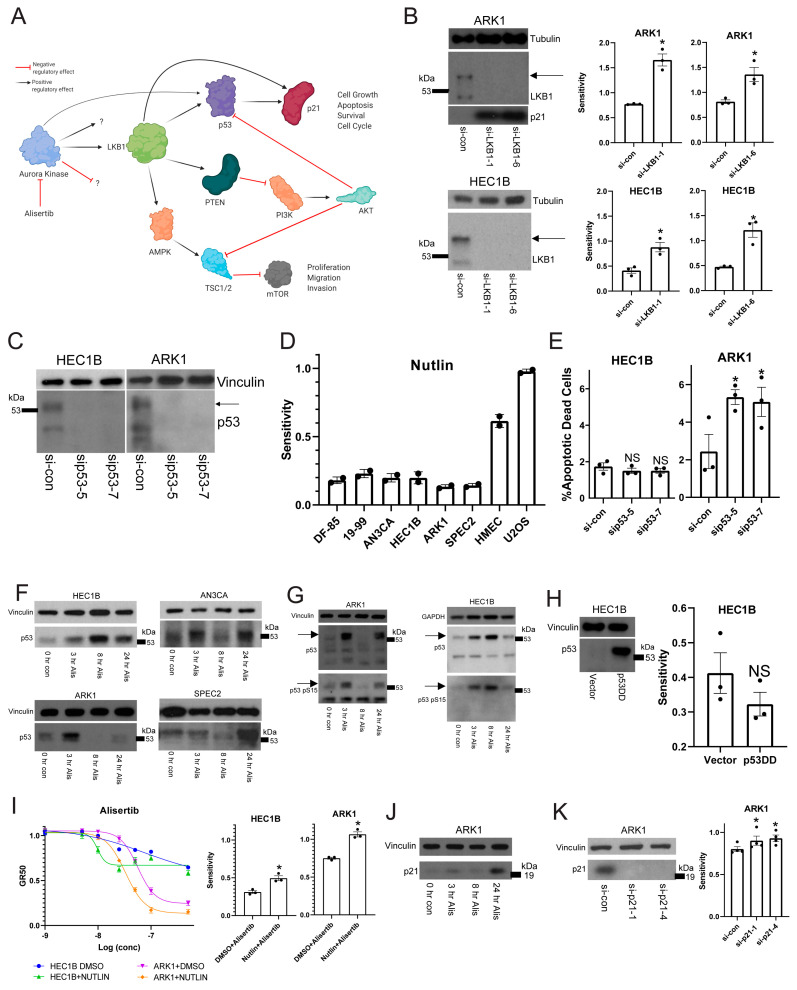
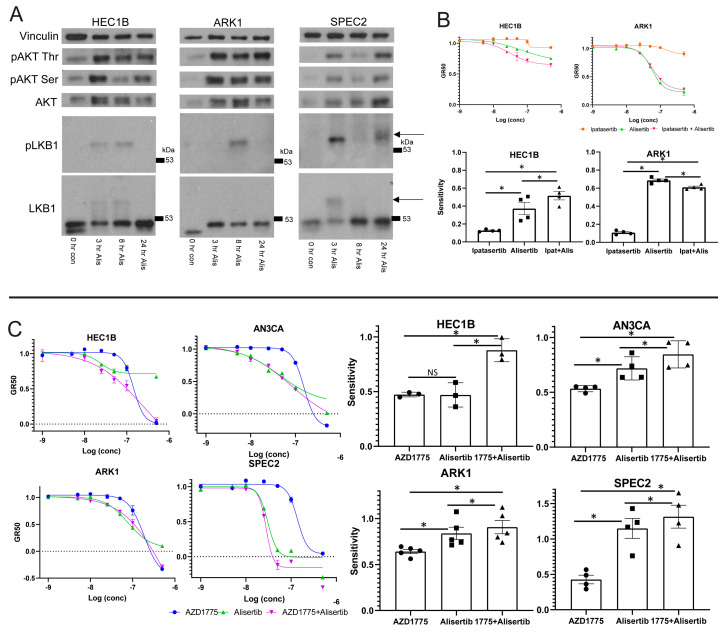
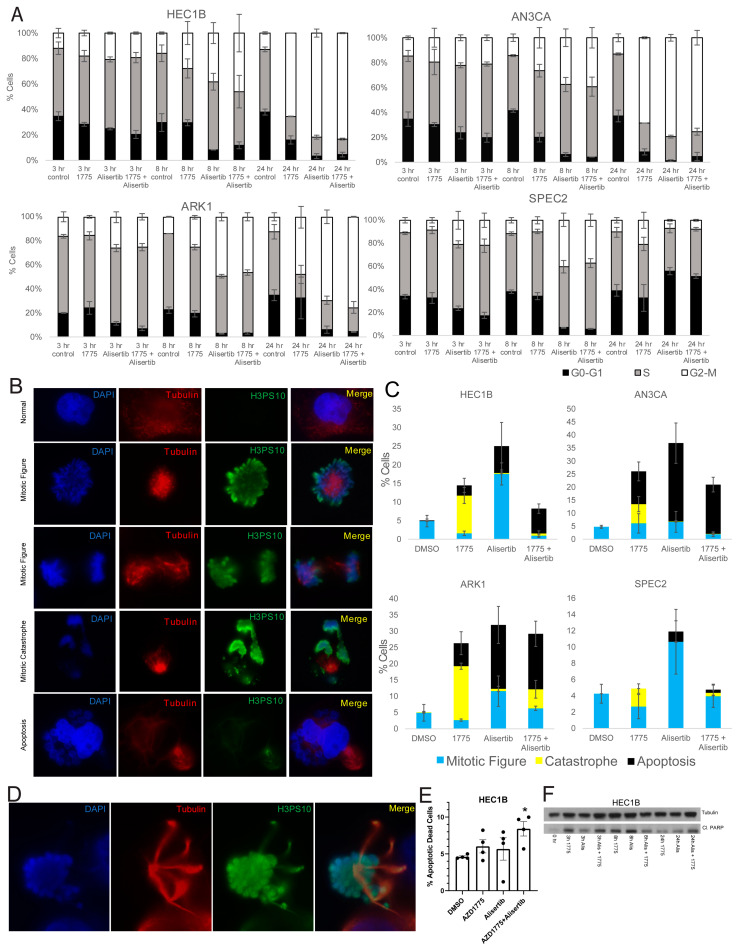
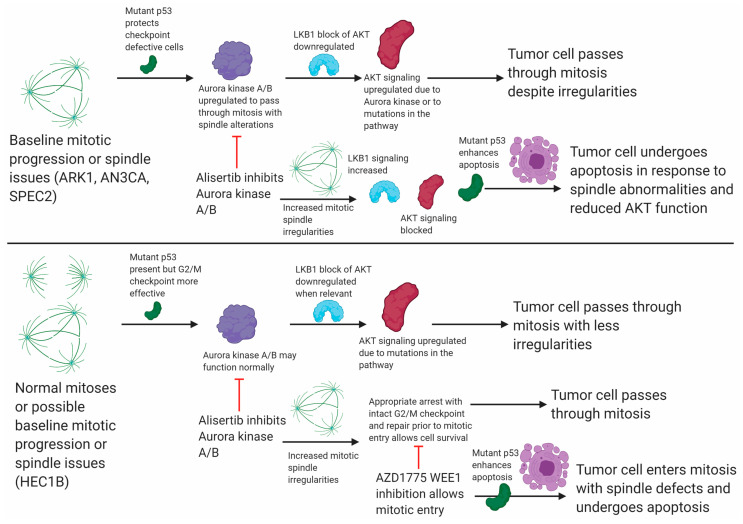
Uterine carcinoma (UC) is the most common gynecologic malignancy in the United States. TP53 mutant UCs cause a disproportionate number of deaths due to limited therapies for these tumors and the lack of mechanistic understanding of their fundamental vulnerabilities. Here we sought to understand the functional and therapeutic relevance of TP53 mutations in UC. We functionally profiled targetable TP53 dependent DNA damage repair and cell cycle control pathways in a panel of TP53 mutant UC cell lines and patient-derived organoids. There were no consistent defects in DNA damage repair pathways. Rather, most models demonstrated dependence on defective G2/M cell cycle checkpoints and subsequent upregulation of Aurora kinase-LKB1-p53-AKT signaling in the setting of baseline mitotic defects. This combination makes them sensitive to Aurora kinase inhibition. Resistant lines demonstrated an intact G2/M checkpoint, and combining Aurora kinase and WEE1 inhibitors, which then push these cells through mitosis with Aurora kinase inhibitor-induced spindle defects, led to apoptosis in these cases. Overall, this work presents Aurora kinase inhibitors alone or in combination with WEE1 inhibitors as relevant mechanism driven therapies for TP53 mutant UCs. Context specific functional assessment of the G2/M checkpoint may serve as a biomarker in identifying Aurora kinase inhibitor sensitive tumors.
Keywords: Aurora kinase; G2/M cell cycle checkpoint; LKB1; p53; uterine cancer.
Conflict of interest statement
U.A.M. is a consultant for Novartis, Merck, and Nextcure, is on the scientific advisory boards for 2X Oncology (unpaid) and the Clearity Foundation, and is an uncompensated advisor for AstraZeneca. S.J.H. receives sponsored research support from Eli Lilly and Company and AstraZeneca not related to this work. B.R.R. is an uncompensated scientific advisory board member for VincenTech. B.R.R. receives sponsored research support from Palleon Pharmaceuticals and Mercy Bioanalytics. Neither is directly related to the current research. J.F.L. is a consultant for Genentech and has participated in advisory boards for AstraZeneca, Clovis, Genentech, Merck, Regeneron Therapeutics, and GlaxoSmithKline, outside the submitted work. J.F.L. reports funding to her institution for study conduct as PI on trials from 2X Oncology, Aravive, Arch Oncology, AstraZeneca, Bristol-Myers Squibb, Clovis Oncology, CytomX Therapeutics, GlaxoSmithKline, Regeneron, Surface Oncology, Tesaro, and Vigeo Therapeutics, outside the submitted work. The remaining authors declare no competing interests.
Figures
Similar articles
-
Functional Profiling of p53 and RB Cell Cycle Regulatory Proficiency Suggests Mechanism-Driven Molecular Stratification in Endometrial Carcinoma.Cancer Res Commun. 2025 Apr 1;5(4):719-742. doi: 10.1158/2767-9764.CRC-24-0028. Cancer Res Commun. 2025. PMID: 39932274 Free PMC article.
-
WEE1 Kinase Inhibitor AZD1775 Has Preclinical Efficacy in LKB1-Deficient Non-Small Cell Lung Cancer.Cancer Res. 2017 Sep 1;77(17):4663-4672. doi: 10.1158/0008-5472.CAN-16-3565. Epub 2017 Jun 26. Cancer Res. 2017. PMID: 28652249
-
Chk1 and Wee1 control genotoxic-stress induced G2-M arrest in melanoma cells.Cell Signal. 2015 May;27(5):951-60. doi: 10.1016/j.cellsig.2015.01.020. Epub 2015 Feb 12. Cell Signal. 2015. PMID: 25683911
-
Aurora kinases and DNA damage response.Mutat Res. 2020 May-Dec;821:111716. doi: 10.1016/j.mrfmmm.2020.111716. Epub 2020 Jul 23. Mutat Res. 2020. PMID: 32738522 Review.
-
Functional Significance of Aurora Kinases-p53 Protein Family Interactions in Cancer.Front Oncol. 2016 Nov 25;6:247. doi: 10.3389/fonc.2016.00247. eCollection 2016. Front Oncol. 2016. PMID: 27933271 Free PMC article. Review.
Cited by
-
Exploiting Cancer Dormancy Signaling Mechanisms in Epithelial Ovarian Cancer Through Spheroid and Organoid Analysis.Cells. 2025 Jan 17;14(2):133. doi: 10.3390/cells14020133. Cells. 2025. PMID: 39851561 Free PMC article.
References
-
- Hamilton C.A., Pothuri B., Arend R.C., Backes F.J., Gehrig P.A., Soliman P.T., Thompson J.S., Urban R.R., Burke W.M. Endometrial cancer: A society of gynecologic oncology evidence-based review and recommendations, part II. Gynecol. Oncol. 2021;160:827–834. doi: 10.1016/j.ygyno.2020.12.024. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
