

First Description of SARS-CoV-2 Infection in Two Feral American Mink (Neovison vison) Caught in the Wild
- PMID: 34065657
- PMCID: PMC8156136
- DOI: 10.3390/ani11051422
First Description of SARS-CoV-2 Infection in Two Feral American Mink (Neovison vison) Caught in the Wild
Abstract
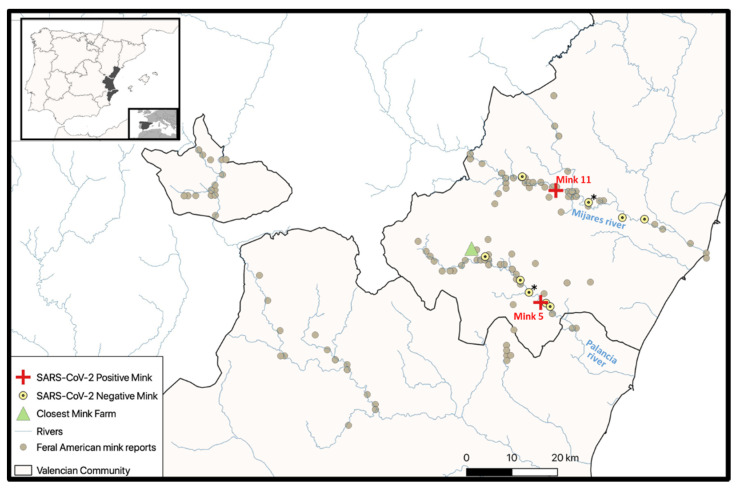
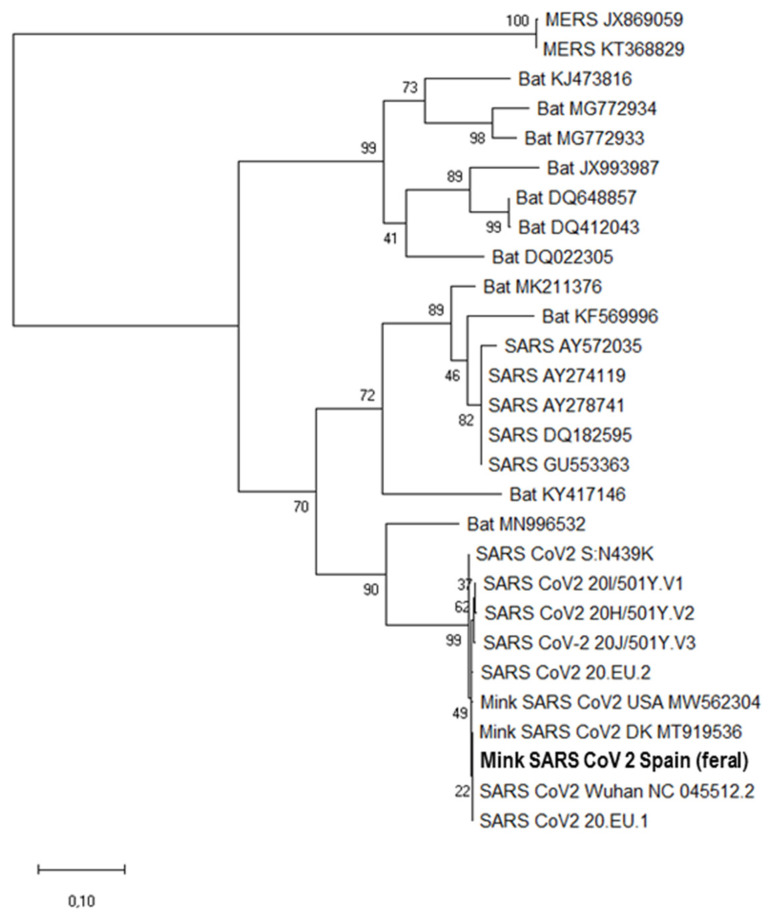
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causal agent of COVID-19, is considered a pathogen of animal origin that is mainly transmitted from human to human. Several animal species can be naturally or experimentally infected by SARS-CoV-2, with compelling evidence that mink is highly susceptible to SARS-CoV-2 infection. Human-to-mink infection cases have been reported and there are also suggestions that mink-to-human infection occurs. Mink infections have been reported to date only on fur farms, except for one infected free- ranging wild mink near a Utah (USA) fur farm, which suggests a transmission pathway from farms to wild mink. We now report the detection of SARS-CoV-2 in 2 of 13 feral dark brown American mink (Neovison vison) trapped in the Valencian Community (Eastern Spain), during an invasive species trapping campaign. They were trapped in riverbeds in sparsely inhabited rural areas known to harbor self-sustained feral mink populations. The closest fur farm is about 20 km away. SARS-CoV-2 RNA was detected by two-step RT-PCR in these animals' mesenteric lymph nodes and was confirmed by sequencing a 397-nucleotide amplified region of the S gene, yielding identical sequences in both animals. A molecular phylogenetic analysis was run on this sequence, which was found to correspond to the consensus SARS-CoV-2 sequence from Wuhan. Our findings appear to represent the first example of SARS-CoV-2 acquired in the wild by feral mink in self-sustained populations.
Keywords: American mink; COVID-19; Neovison vison; SARS-CoV-2; spike; wildlife.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
