

Neurons and Glia Interplay in α-Synucleinopathies
- PMID: 34066733
- PMCID: PMC8125822
- DOI: 10.3390/ijms22094994
Neurons and Glia Interplay in α-Synucleinopathies
Abstract
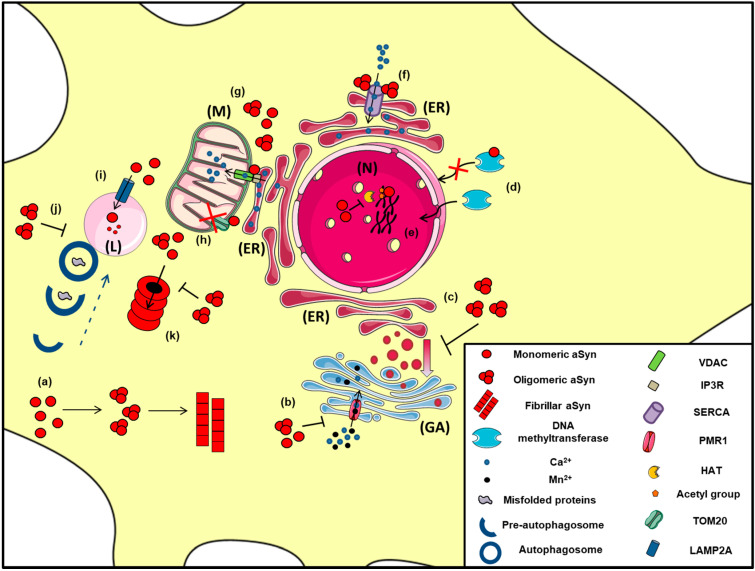
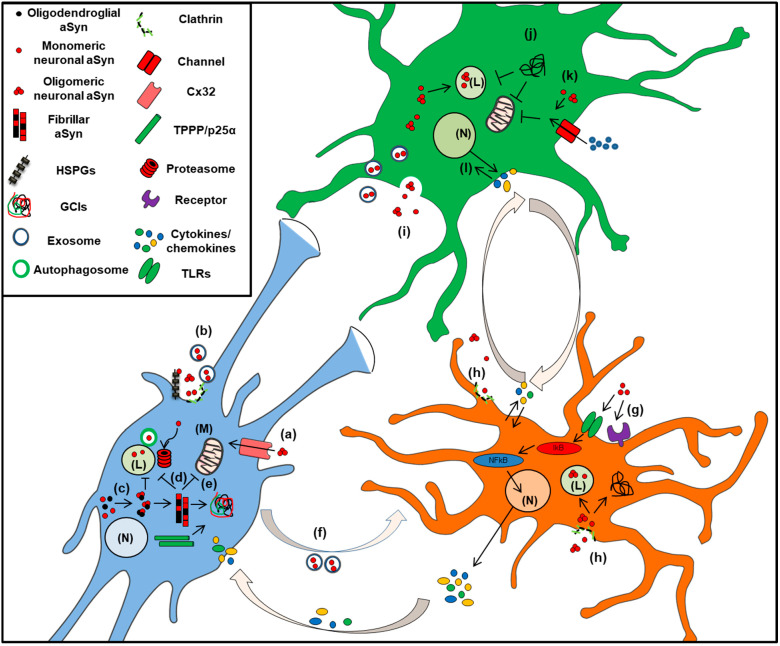
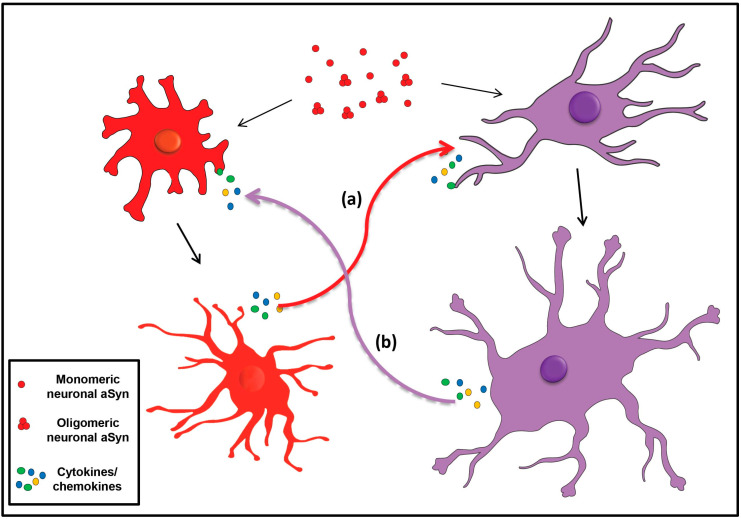
Accumulation of the neuronal presynaptic protein alpha-synuclein within proteinaceous inclusions represents the key histophathological hallmark of a spectrum of neurodegenerative disorders, referred to by the umbrella term a-synucleinopathies. Even though alpha-synuclein is expressed predominantly in neurons, pathological aggregates of the protein are also found in the glial cells of the brain. In Parkinson's disease and dementia with Lewy bodies, alpha-synuclein accumulates mainly in neurons forming the Lewy bodies and Lewy neurites, whereas in multiple system atrophy, the protein aggregates mostly in the glial cytoplasmic inclusions within oligodendrocytes. In addition, astrogliosis and microgliosis are found in the synucleinopathy brains, whereas both astrocytes and microglia internalize alpha-synuclein and contribute to the spread of pathology. The mechanisms underlying the pathological accumulation of alpha-synuclein in glial cells that under physiological conditions express low to non-detectable levels of the protein are an area of intense research. Undoubtedly, the presence of aggregated alpha-synuclein can disrupt glial function in general and can contribute to neurodegeneration through numerous pathways. Herein, we summarize the current knowledge on the role of alpha-synuclein in both neurons and glia, highlighting the contribution of the neuron-glia connectome in the disease initiation and progression, which may represent potential therapeutic target for a-synucleinopathies.
Keywords: a-Synuclein; aggregation; astrocytes; inclusions; microglia; neurons; oligodendroglia; seeding.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
