

CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings
- PMID: 34068417
- PMCID: PMC8153625
- DOI: 10.3390/ijms22105180
CACNA1A Mutations Causing Early Onset Ataxia: Profiling Clinical, Dysmorphic and Structural-Functional Findings
Abstract
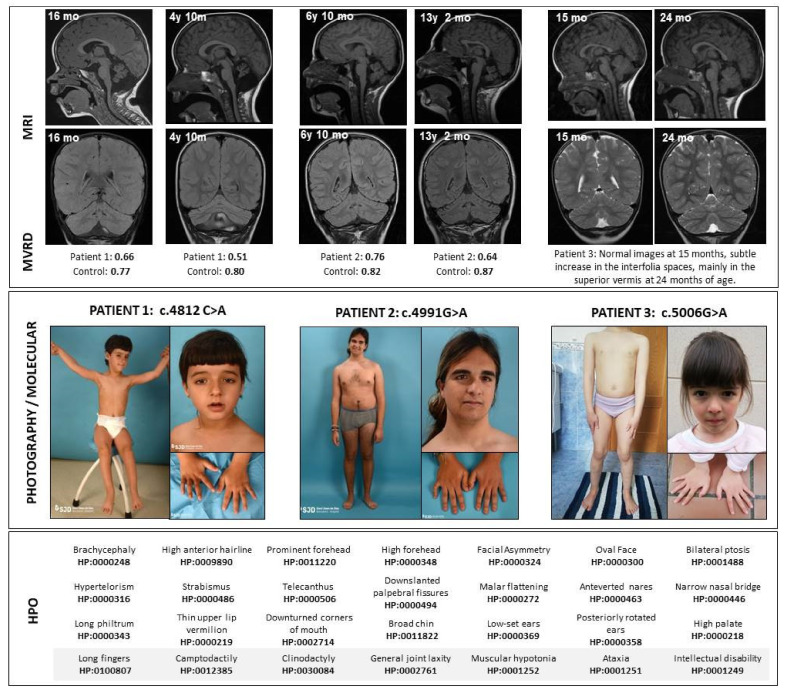
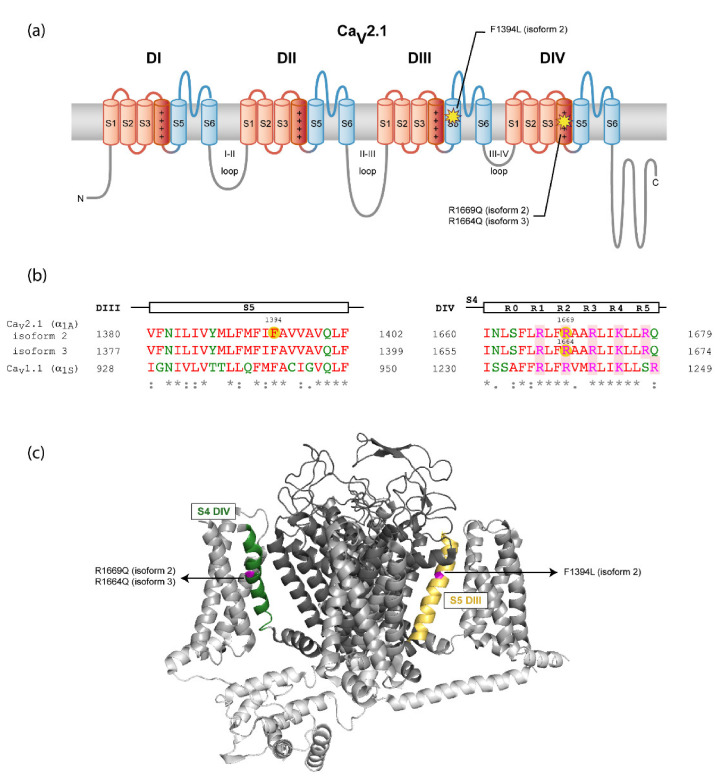
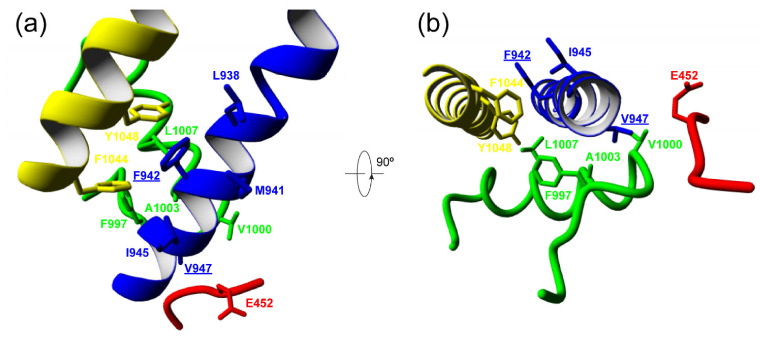
The CACNA1A gene encodes the pore-forming α1A subunit of the voltage-gated CaV2.1 Ca2+ channel, essential in neurotransmission, especially in Purkinje cells. Mutations in CACNA1A result in great clinical heterogeneity with progressive symptoms, paroxysmal events or both. During infancy, clinical and neuroimaging findings may be unspecific, and no dysmorphic features have been reported. We present the clinical, radiological and evolutionary features of three patients with congenital ataxia, one of them carrying a new variant. We report the structural localization of variants and their expected functional consequences. There was an improvement in cerebellar syndrome over time despite a cerebellar atrophy progression, inconsistent response to acetazolamide and positive response to methylphenidate. The patients shared distinctive facial gestalt: oval face, prominent forehead, hypertelorism, downslanting palpebral fissures and narrow nasal bridge. The two α1A affected residues are fully conserved throughout evolution and among the whole human CaV channel family. They contribute to the channel pore and the voltage sensor segment. According to structural data analysis and available functional characterization, they are expected to exert gain- (F1394L) and loss-of-function (R1664Q/R1669Q) effect, respectively. Among the CACNA1A-related phenotypes, our results suggest that non-progressive congenital ataxia is associated with developmental delay and dysmorphic features, constituting a recognizable syndromic neurodevelopmental disorder.
Keywords: CACNA1A gene; CaV2.1 (P/Q-type) voltage-dependent calcium channel; ataxia; cerebellar atrophy; dysmorphic traits; early-onset cerebellar ataxia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Calandriello L., Veneziano L., Francia A., Sabbadini G., Colonnese C., Mantuano E., Jodice C., Trettel F., Viviani P., Manfredi M., et al. Acetazolamide-responsive episodic ataxia in an Italian family refines gene mapping on chromosome 19p13. Brain. 1997;120:805–812. doi: 10.1093/brain/120.5.805. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
