

Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart
- PMID: 34068508
- PMCID: PMC8125982
- DOI: 10.3390/ijms22095025
Dystrophin Deficiency Causes Progressive Depletion of Cardiovascular Progenitor Cells in the Heart
Abstract
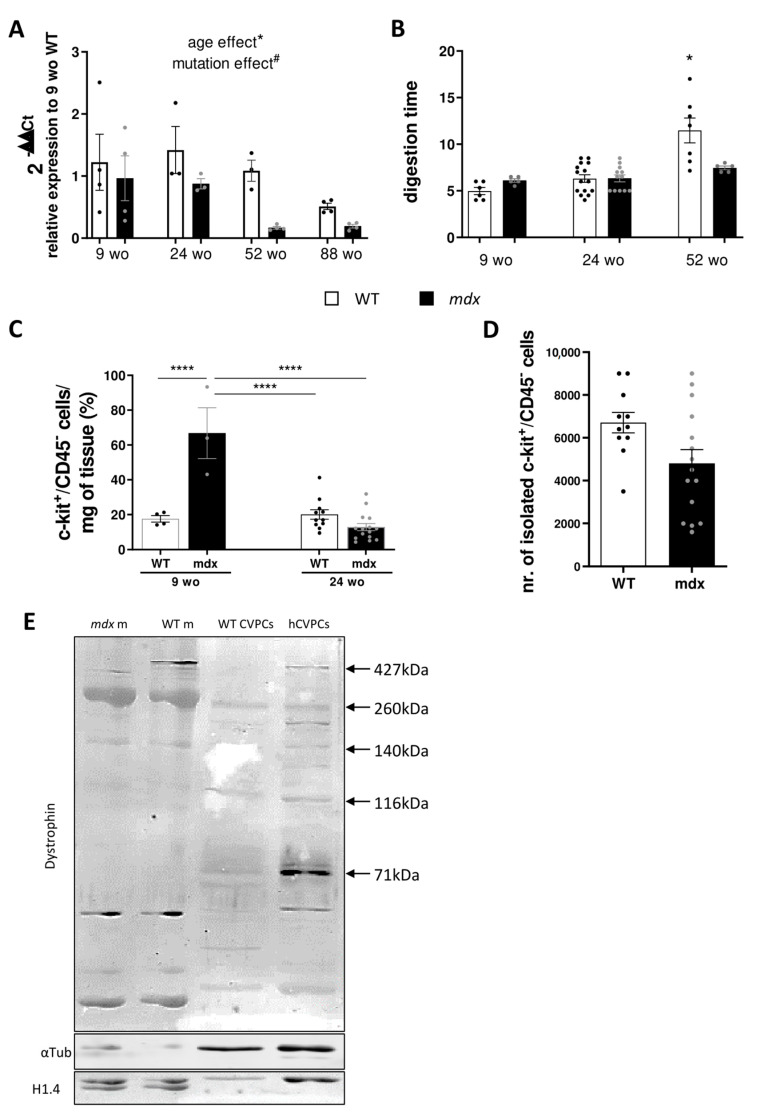
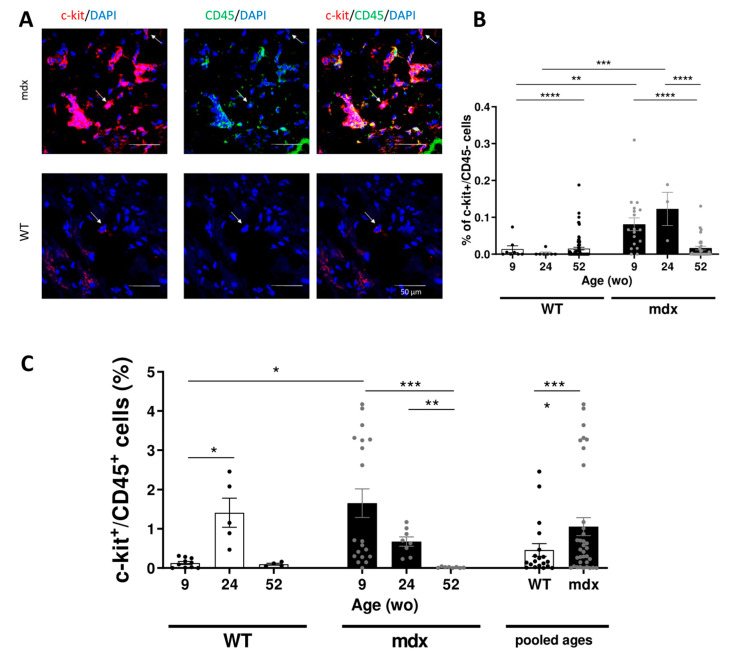
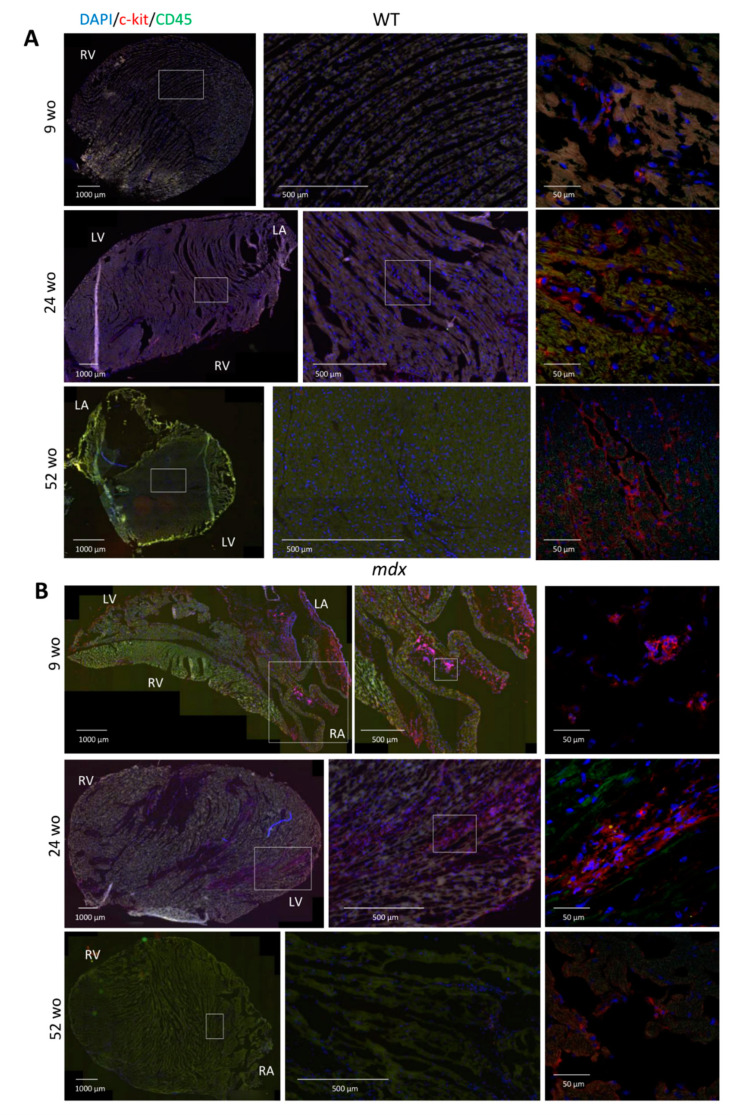
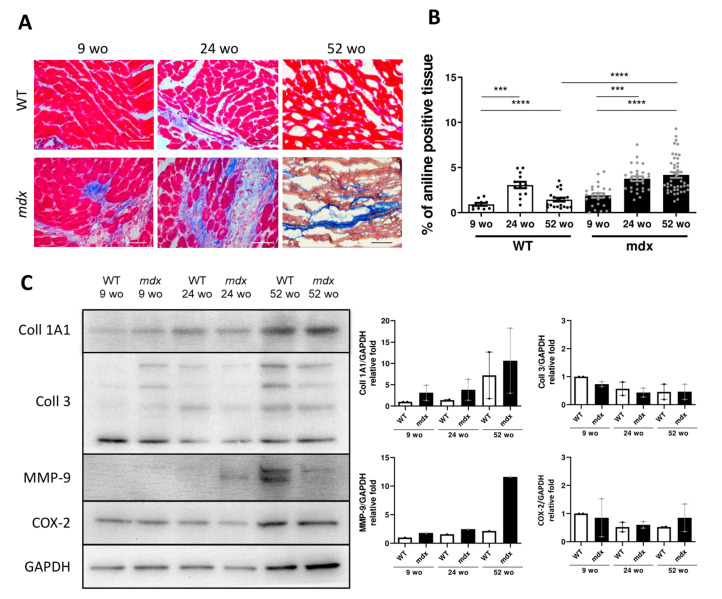
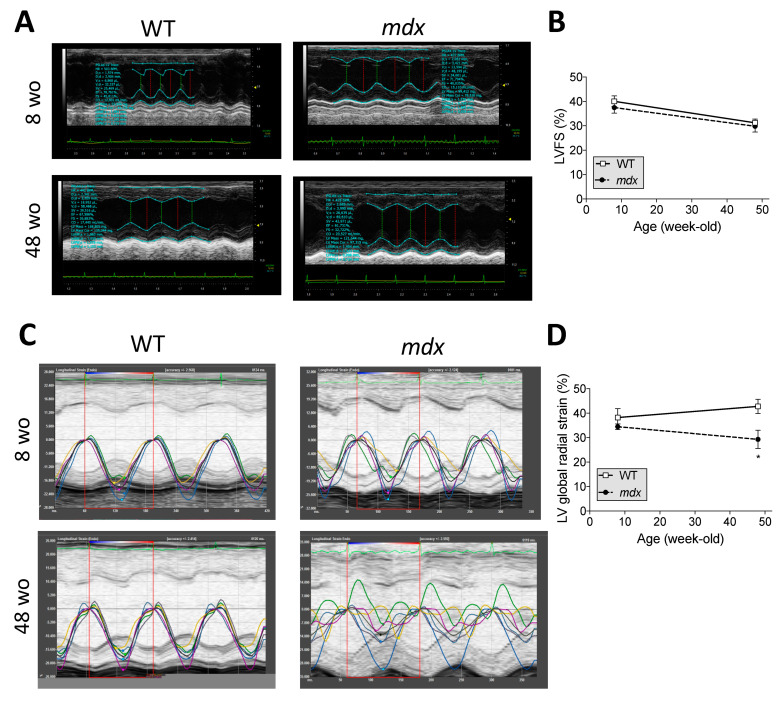
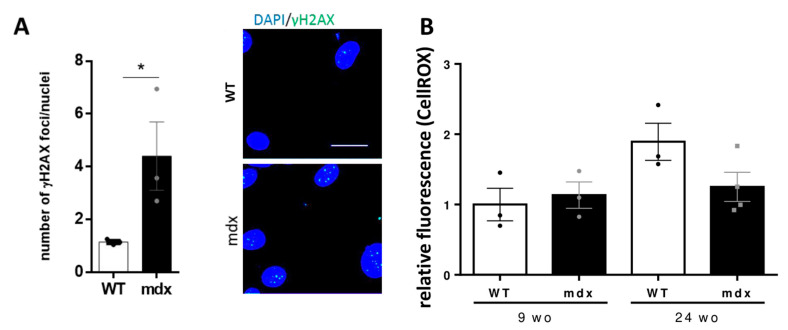
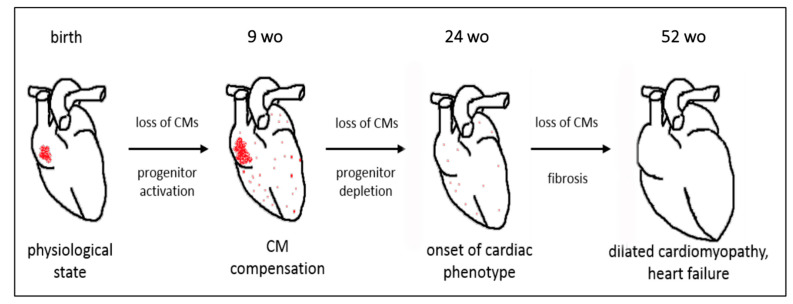
Duchenne muscular dystrophy (DMD) is a devastating condition shortening the lifespan of young men. DMD patients suffer from age-related dilated cardiomyopathy (DCM) that leads to heart failure. Several molecular mechanisms leading to cardiomyocyte death in DMD have been described. However, the pathological progression of DMD-associated DCM remains unclear. In skeletal muscle, a dramatic decrease in stem cells, so-called satellite cells, has been shown in DMD patients. Whether similar dysfunction occurs with cardiac muscle cardiovascular progenitor cells (CVPCs) in DMD remains to be explored. We hypothesized that the number of CVPCs decreases in the dystrophin-deficient heart with age and disease state, contributing to DCM progression. We used the dystrophin-deficient mouse model (mdx) to investigate age-dependent CVPC properties. Using quantitative PCR, flow cytometry, speckle tracking echocardiography, and immunofluorescence, we revealed that young mdx mice exhibit elevated CVPCs. We observed a rapid age-related CVPC depletion, coinciding with the progressive onset of cardiac dysfunction. Moreover, mdx CVPCs displayed increased DNA damage, suggesting impaired cardiac muscle homeostasis. Overall, our results identify the early recruitment of CVPCs in dystrophic hearts and their fast depletion with ageing. This latter depletion may participate in the fibrosis development and the acceleration onset of the cardiomyopathy.
Keywords: c-kit; cardiovascular progenitors; dilated cardiomyopathy; duchenne muscular dystrophy; genomic instability; mdx mouse.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Injection of vessel-derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn-/- but not aged mdx mouse models for duchenne muscular dystrophy.Stem Cells Transl Med. 2013 Jan;2(1):68-80. doi: 10.5966/sctm.2012-0107. Epub 2012 Dec 27. Stem Cells Transl Med. 2013. PMID: 23283493 Free PMC article.
-
Cardiovascular phenotype of the Dmdmdx rat - a suitable animal model for Duchenne muscular dystrophy.Dis Model Mech. 2021 Feb 22;14(2):dmm047704. doi: 10.1242/dmm.047704. Dis Model Mech. 2021. PMID: 33619211 Free PMC article.
-
Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart.Neuromuscul Disord. 2008 May;18(5):371-81. doi: 10.1016/j.nmd.2008.03.008. Epub 2008 Apr 25. Neuromuscul Disord. 2008. PMID: 18440230 Free PMC article.
-
Modeling Duchenne Muscular Dystrophy Cardiomyopathy with Patients' Induced Pluripotent Stem-Cell-Derived Cardiomyocytes.Int J Mol Sci. 2023 May 12;24(10):8657. doi: 10.3390/ijms24108657. Int J Mol Sci. 2023. PMID: 37240001 Free PMC article. Review.
-
Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies.J Mol Cell Cardiol. 2013 May;58:217-24. doi: 10.1016/j.yjmcc.2012.12.009. Epub 2012 Dec 20. J Mol Cell Cardiol. 2013. PMID: 23261966 Free PMC article. Review.
Cited by
-
Evaluation of Cardiac Function in Young Mdx Mice Using MRI with Feature Tracking and Self-Gated Magnetic Resonance Cine Imaging.Diagnostics (Basel). 2023 Apr 19;13(8):1472. doi: 10.3390/diagnostics13081472. Diagnostics (Basel). 2023. PMID: 37189573 Free PMC article.
References
-
- Amedro P., Vincenti M., De La Villeon G., Lavastre K., Barrea C., Guillaumont S., Bredy C., Gamon L., Meli A.C., Cazorla O., et al. Speckle-Tracking Echocardiography in Children with Duchenne Muscular Dystrophy: A Prospective Multicenter Controlled Cross-Sectional Study. J. Am. Soc. Echocardiogr. 2019;32:412–422. doi: 10.1016/j.echo.2018.10.017. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
