

A Large Family with p.Arg554His Mutation in ABCD1: Clinical Features and Genotype/Phenotype Correlation in Female Carriers
- PMID: 34069712
- PMCID: PMC8160645
- DOI: 10.3390/genes12050775
A Large Family with p.Arg554His Mutation in ABCD1: Clinical Features and Genotype/Phenotype Correlation in Female Carriers
Abstract
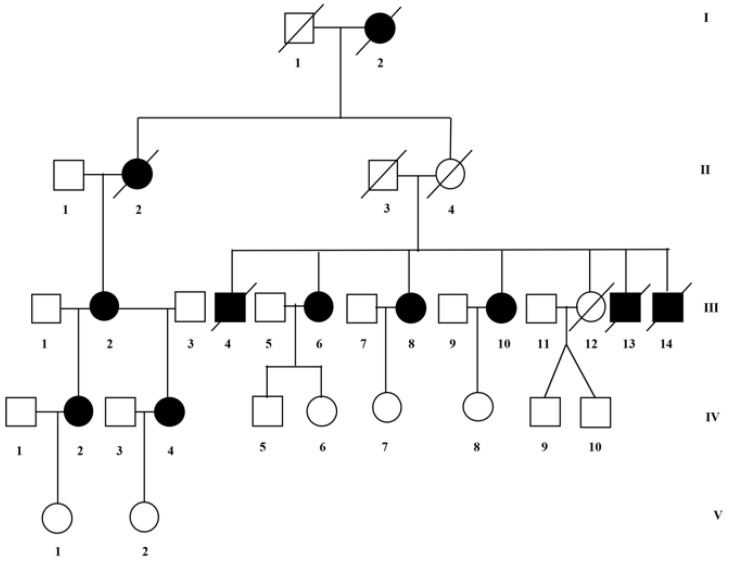
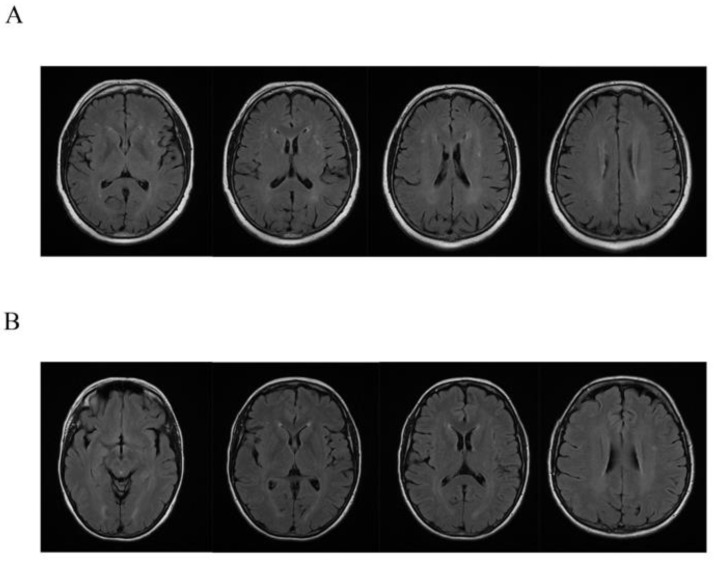
X-linked adrenoleukodystrophy (X-ALD, OMIM #300100) is the most common peroxisomal disorder clinically characterized by two main phenotypes: adrenomyeloneuropathy (AMN) and the cerebral demyelinating form of X-ALD (cerebral ALD). The disease is caused by defects in the gene for the adenosine triphosphate (ATP)-binding cassette protein, subfamily D (ABCD1) that encodes the peroxisomal transporter of very-long-chain fatty acids (VLCFAs). The defective function of ABCD1 protein prevents β-oxidation of VLCFAs, which thus accumulate in tissues and plasma, to represent the hallmark of the disease. As in many X-linked diseases, it has been routinely expected that female carriers are asymptomatic. Nonetheless, recent findings indicate that most ABCD1 female carriers become symptomatic, with a motor disability that typically appears between the fourth and fifth decade. In this paper, we report a large family in which affected males died during the first decade, while affected females develop, during the fourth decade, progressive lower limb weakness with spastic or ataxic-spastic gait, tetra-hyperreflexia with sensory alterations. Clinical and genetic evaluations were performed in nine subjects, eight females (five affected and three healthy) and one healthy male. All affected females were carriers of the c.1661G>A (p.Arg554His, rs201568579) mutation. This study strengthens the relevance of clinical symptoms in female carriers of ABCD1 mutations, which leads to a better understanding of the role of the genetic background and the genotype-phenotype correlation. This indicates the relevance to include ABCD1 genes in genetic panels for gait disturbance in women.
Keywords: ABCD1; X-linked adrenoleukodystrophy; diagnosis; neurogenetics; next generation sequencing.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Investigating ABCD1 mutations in a Taiwanese cohort with hereditary spastic paraplegia phenotype.Parkinsonism Relat Disord. 2021 Nov;92:7-12. doi: 10.1016/j.parkreldis.2021.10.006. Epub 2021 Oct 9. Parkinsonism Relat Disord. 2021. PMID: 34649108
-
[Screening for carrier and prenatal diagnosis of X-linked adrenoleukodystrophy].Zhonghua Er Ke Za Zhi. 2005 May;43(5):345-9. Zhonghua Er Ke Za Zhi. 2005. PMID: 15924749 Chinese.
-
ABCD1 translation-initiator mutation demonstrates genotype-phenotype correlation for AMN.Neurology. 2001 Dec 11;57(11):1956-62. doi: 10.1212/wnl.57.11.1956. Neurology. 2001. PMID: 11739809
-
[X-linked adrenoleukodystrophy].Ann Endocrinol (Paris). 2007 Dec;68(6):403-11. doi: 10.1016/j.ando.2007.04.002. Epub 2007 May 29. Ann Endocrinol (Paris). 2007. PMID: 17532287 Review. French.
-
Management of adrenoleukodystrophy: From pre-clinical studies to the development of new therapies.Biomed Pharmacother. 2021 Nov;143:112214. doi: 10.1016/j.biopha.2021.112214. Epub 2021 Sep 21. Biomed Pharmacother. 2021. PMID: 34560537 Review.
Cited by
-
X-linked adrenoleukodystrophy caused by maternal ABCD1 mutation and paternal X chromosome inactivation.Exp Ther Med. 2022 Jul 12;24(3):565. doi: 10.3892/etm.2022.11502. eCollection 2022 Sep. Exp Ther Med. 2022. PMID: 35978942 Free PMC article.
-
Short-read genome sequencing allows 'en route' diagnosis of patients with atypical Friedreich ataxia.J Neurol. 2023 Aug;270(8):4112-4117. doi: 10.1007/s00415-023-11745-8. Epub 2023 Apr 29. J Neurol. 2023. PMID: 37119371 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
