

Insulin Resistance and Diabetes Mellitus in Alzheimer's Disease
- PMID: 34069890
- PMCID: PMC8157600
- DOI: 10.3390/cells10051236
Insulin Resistance and Diabetes Mellitus in Alzheimer's Disease
Abstract
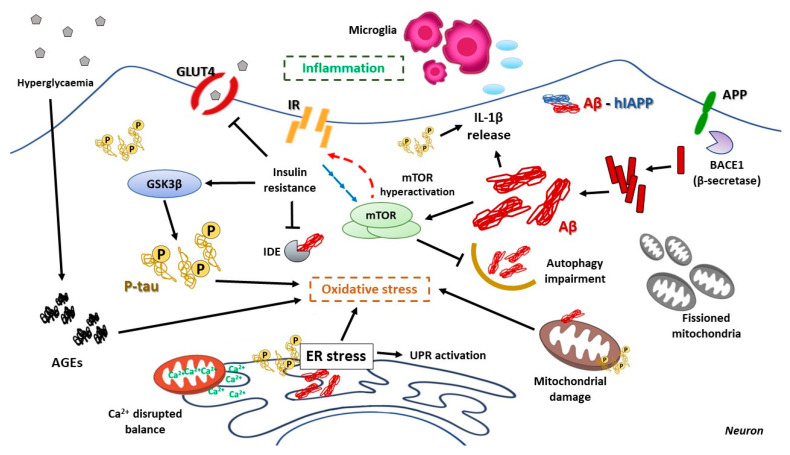
Type 2 diabetes mellitus is a progressive disease that is characterized by the appearance of insulin resistance. The term insulin resistance is very wide and could affect different proteins involved in insulin signaling, as well as other mechanisms. In this review, we have analyzed the main molecular mechanisms that could be involved in the connection between type 2 diabetes and neurodegeneration, in general, and more specifically with the appearance of Alzheimer's disease. We have studied, in more detail, the different processes involved, such as inflammation, endoplasmic reticulum stress, autophagy, and mitochondrial dysfunction.
Keywords: ER stress; T3DM; autophagy; inflammation; insulin resistance; mTOR.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
