

Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations
- PMID: 34070175
- PMCID: PMC8158508
- DOI: 10.3390/v13050936
Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations
Abstract
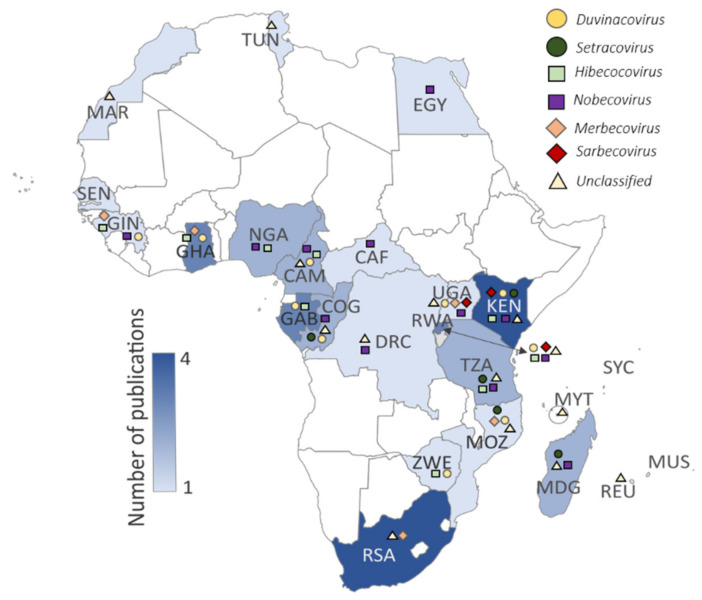
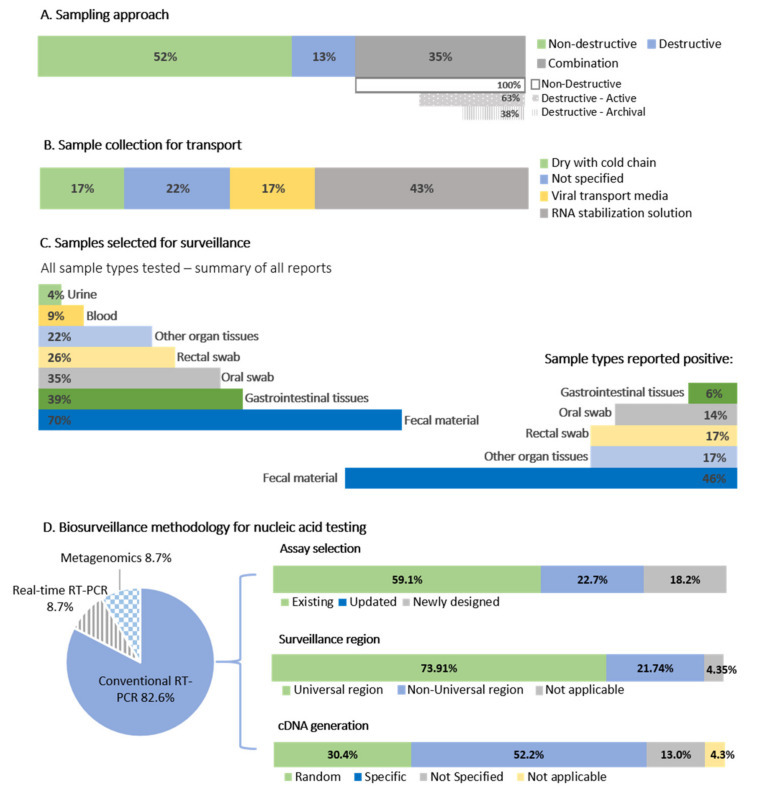
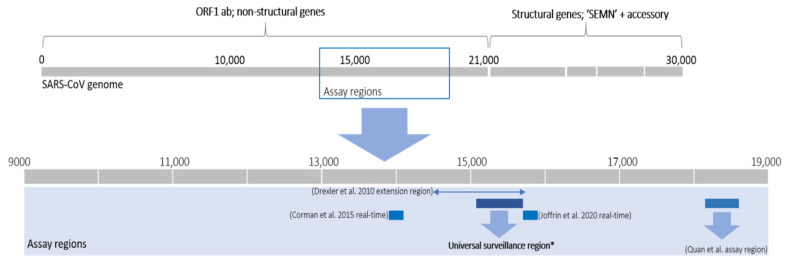
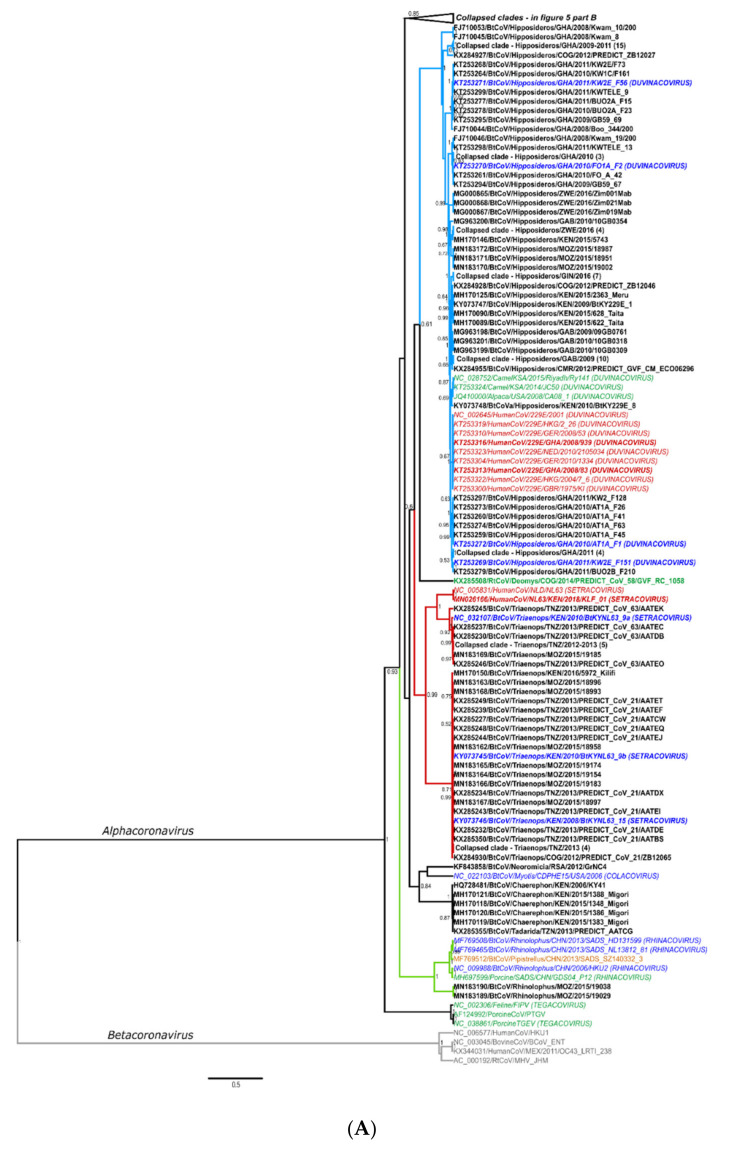
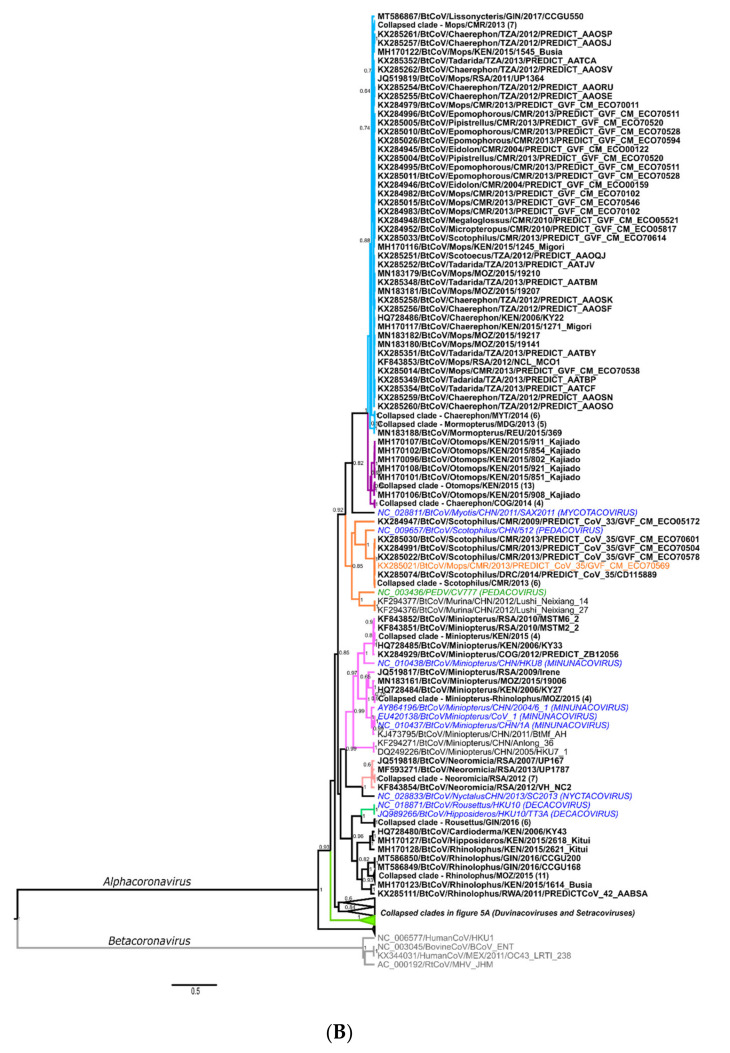
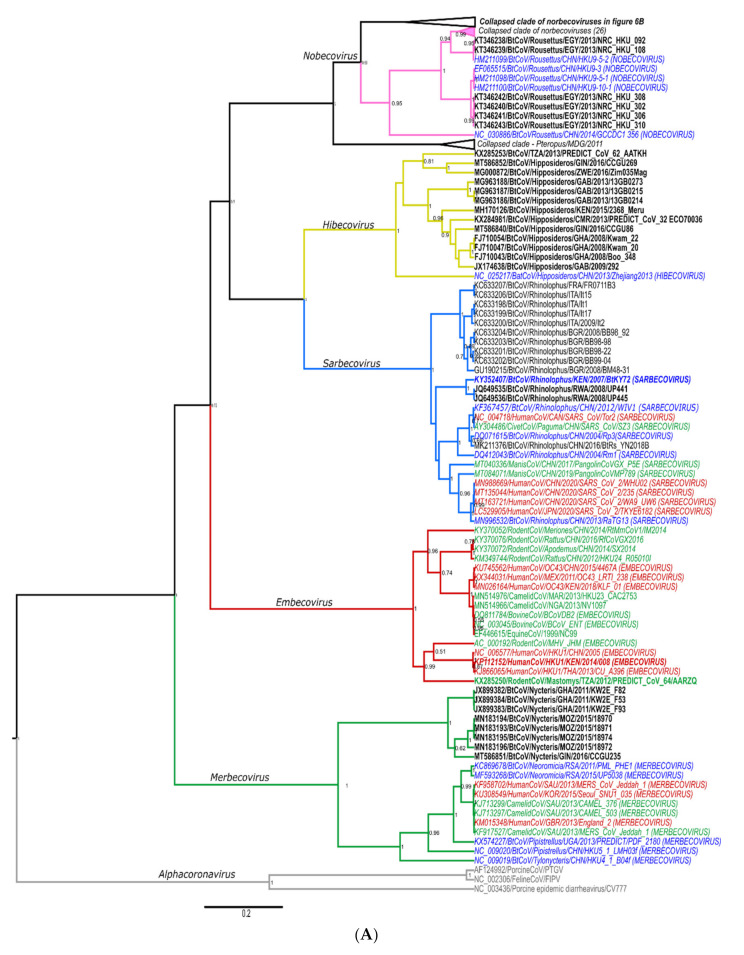
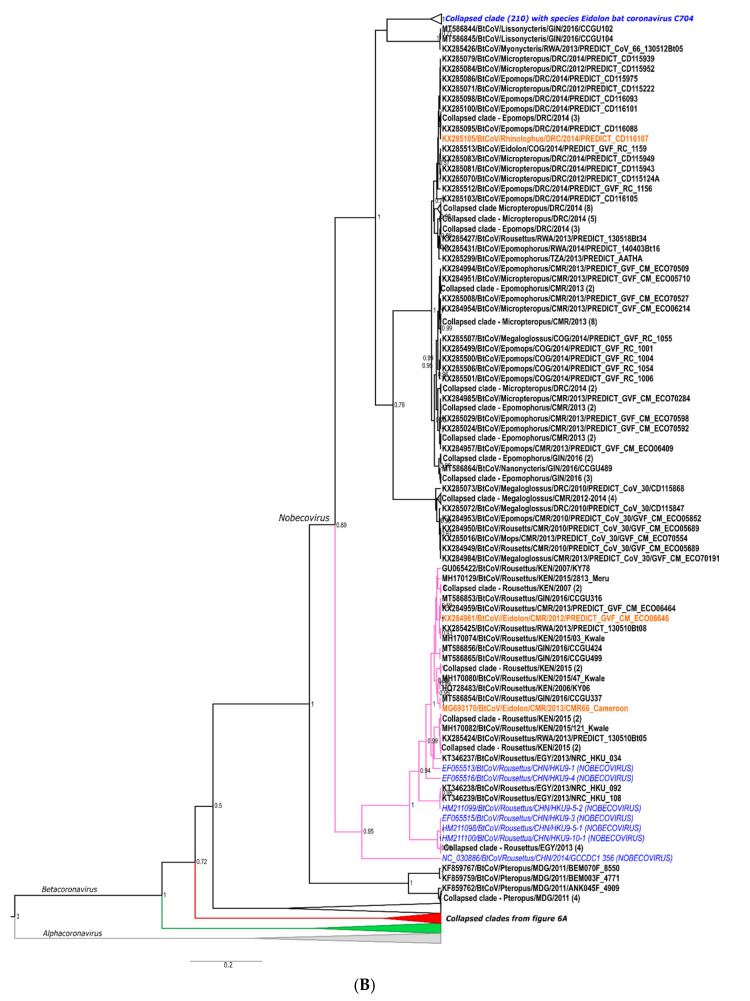
The ongoing coronavirus disease 2019 (COVID-19) pandemic has had devastating health and socio-economic impacts. Human activities, especially at the wildlife interphase, are at the core of forces driving the emergence of new viral agents. Global surveillance activities have identified bats as the natural hosts of diverse coronaviruses, with other domestic and wildlife animal species possibly acting as intermediate or spillover hosts. The African continent is confronted by several factors that challenge prevention and response to novel disease emergences, such as high species diversity, inadequate health systems, and drastic social and ecosystem changes. We reviewed published animal coronavirus surveillance studies conducted in Africa, specifically summarizing surveillance approaches, species numbers tested, and findings. Far more surveillance has been initiated among bat populations than other wildlife and domestic animals, with nearly 26,000 bat individuals tested. Though coronaviruses have been identified from approximately 7% of the total bats tested, surveillance among other animals identified coronaviruses in less than 1%. In addition to a large undescribed diversity, sequences related to four of the seven human coronaviruses have been reported from African bats. The review highlights research gaps and the disparity in surveillance efforts between different animal groups (particularly potential spillover hosts) and concludes with proposed strategies for improved future biosurveillance.
Keywords: Africa; African bat coronaviruses; COVID-19; HCoV-229E; HCoV-NL63; MERS-CoV; SARS-CoV; SARS-CoV 2; bat; biosurveillance; coronaviruses; domestic animals; emerging; surveillance; surveillance strategies; wildlife.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures

Similar articles
-
Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History.J Virol. 2017 Feb 14;91(5):e01953-16. doi: 10.1128/JVI.01953-16. Print 2017 Mar 1. J Virol. 2017. PMID: 28077633 Free PMC article.
-
Properties of Coronavirus and SARS-CoV-2.Malays J Pathol. 2020 Apr;42(1):3-11. Malays J Pathol. 2020. PMID: 32342926 Review.
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
-
Evidence for an Ancestral Association of Human Coronavirus 229E with Bats.J Virol. 2015 Dec;89(23):11858-70. doi: 10.1128/JVI.01755-15. Epub 2015 Sep 16. J Virol. 2015. PMID: 26378164 Free PMC article.
-
Bat origin of human coronaviruses.Virol J. 2015 Dec 22;12:221. doi: 10.1186/s12985-015-0422-1. Virol J. 2015. PMID: 26689940 Free PMC article. Review.
Cited by
-
Advances in understanding bat infection dynamics across biological scales.Proc Biol Sci. 2024 Mar 13;291(2018):20232823. doi: 10.1098/rspb.2023.2823. Epub 2024 Mar 6. Proc Biol Sci. 2024. PMID: 38444339 Free PMC article. Review.
-
Seasonal shedding of coronavirus by straw-colored fruit bats at urban roosts in Africa.PLoS One. 2022 Sep 15;17(9):e0274490. doi: 10.1371/journal.pone.0274490. eCollection 2022. PLoS One. 2022. PMID: 36107832 Free PMC article.
-
Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats.Elife. 2022 Nov 8;11:e79777. doi: 10.7554/eLife.79777. Elife. 2022. PMID: 36346652 Free PMC article.
-
Study of coronavirus diversity in wildlife in Northern Cambodia suggests continuous circulation of SARS-CoV-2-related viruses in bats.Sci Rep. 2025 Apr 12;15(1):12628. doi: 10.1038/s41598-025-92475-x. Sci Rep. 2025. PMID: 40221475 Free PMC article.
-
Lack of detection of SARS-CoV-2 in wildlife from Kerala, India in 2020-21.Access Microbiol. 2024 Jan 31;6(1):000686.v3. doi: 10.1099/acmi.0.000686.v3. eCollection 2024. Access Microbiol. 2024. PMID: 38361659 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
