

Mitochondrial Ca2+ Signaling in Health, Disease and Therapy
- PMID: 34070562
- PMCID: PMC8230075
- DOI: 10.3390/cells10061317
Mitochondrial Ca2+ Signaling in Health, Disease and Therapy
Abstract
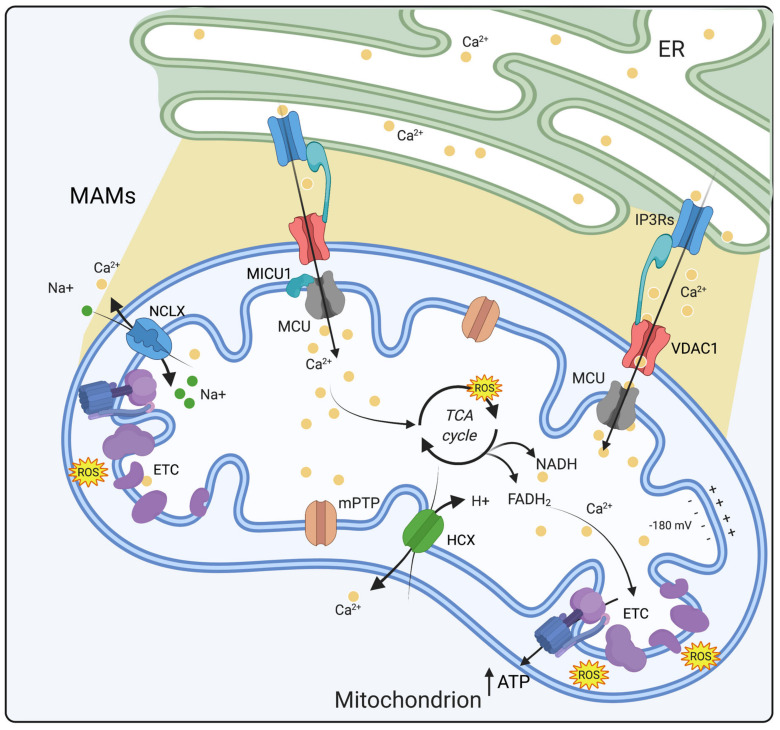
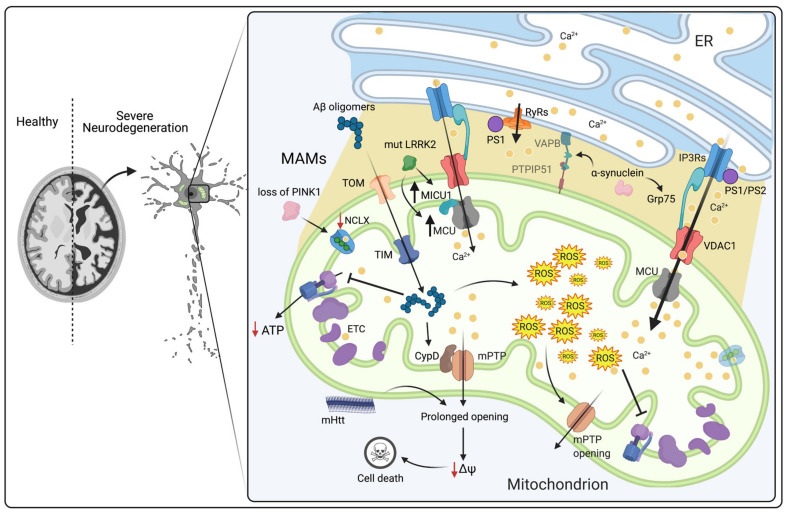
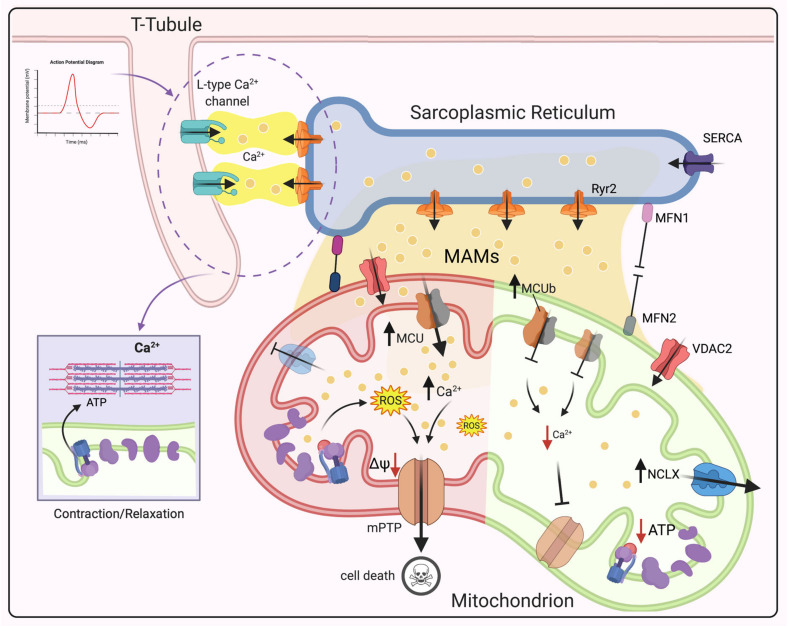
The divalent cation calcium (Ca2+) is considered one of the main second messengers inside cells and acts as the most prominent signal in a plethora of biological processes. Its homeostasis is guaranteed by an intricate and complex system of channels, pumps, and exchangers. In this context, by regulating cellular Ca2+ levels, mitochondria control both the uptake and release of Ca2+. Therefore, at the mitochondrial level, Ca2+ plays a dual role, participating in both vital physiological processes (ATP production and regulation of mitochondrial metabolism) and pathophysiological processes (cell death, cancer progression and metastasis). Hence, it is not surprising that alterations in mitochondrial Ca2+ (mCa2+) pathways or mutations in Ca2+ transporters affect the activities and functions of the entire cell. Indeed, it is widely recognized that dysregulation of mCa2+ signaling leads to various pathological scenarios, including cancer, neurological defects and cardiovascular diseases (CVDs). This review summarizes the current knowledge on the regulation of mCa2+ homeostasis, the related mechanisms and the significance of this regulation in physiology and human diseases. We also highlight strategies aimed at remedying mCa2+ dysregulation as promising therapeutical approaches.
Keywords: Ca2+; cancer; cardiovascular diseases; mPTP; mitochondria; neurodegenerative diseases; therapy.
Conflict of interest statement
The authors declare no conflict of interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
