

Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect
- PMID: 34070744
- PMCID: PMC8197790
- DOI: 10.3390/ijms22115859
Regulation of ClC-2 Chloride Channel Proteostasis by Molecular Chaperones: Correction of Leukodystrophy-Associated Defect
Abstract
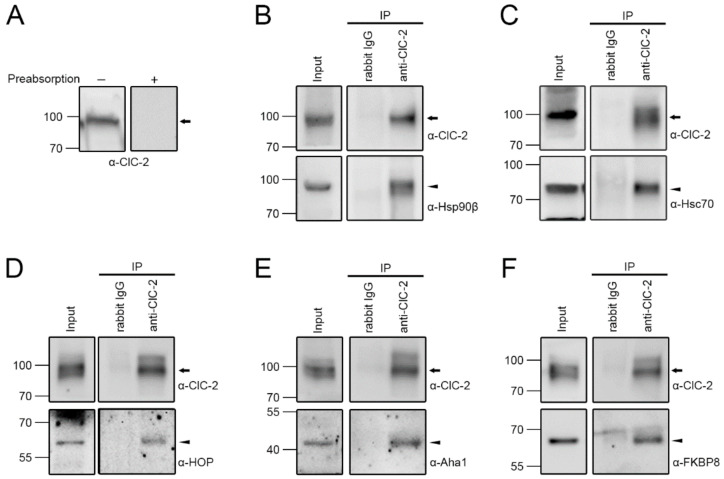
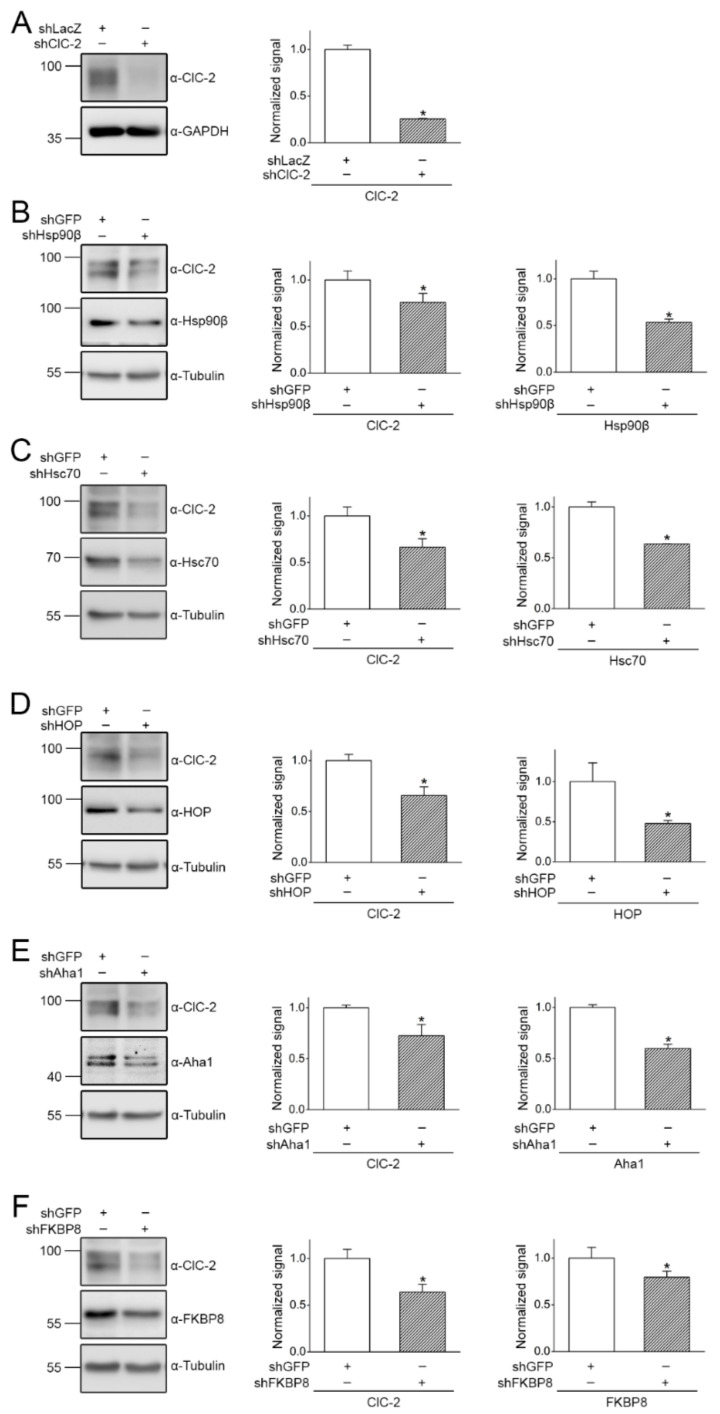
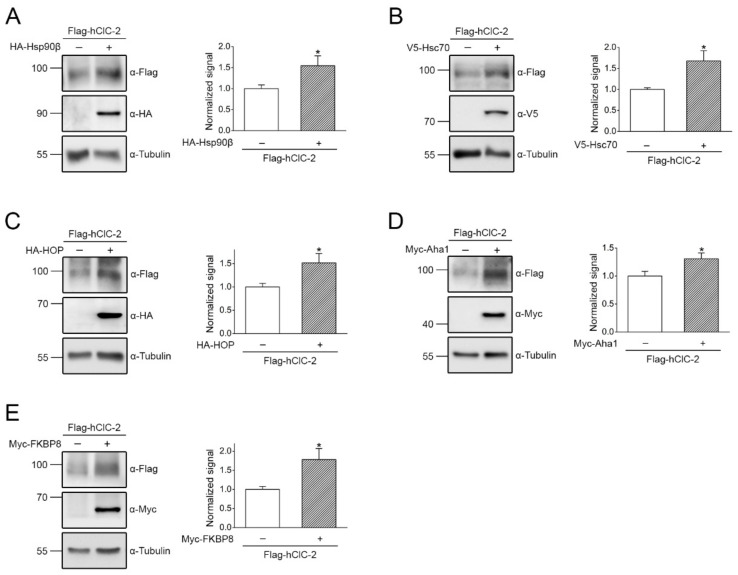
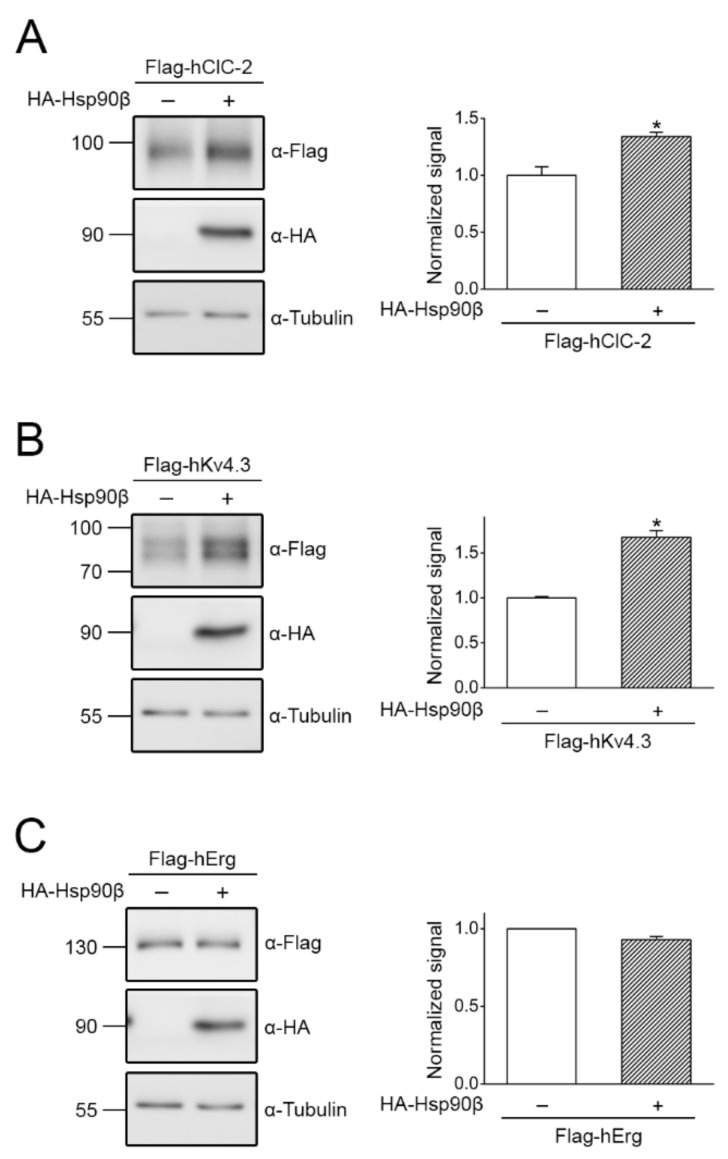
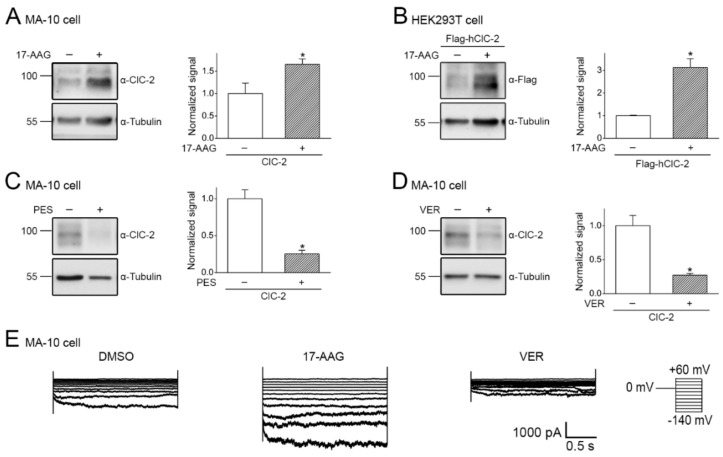
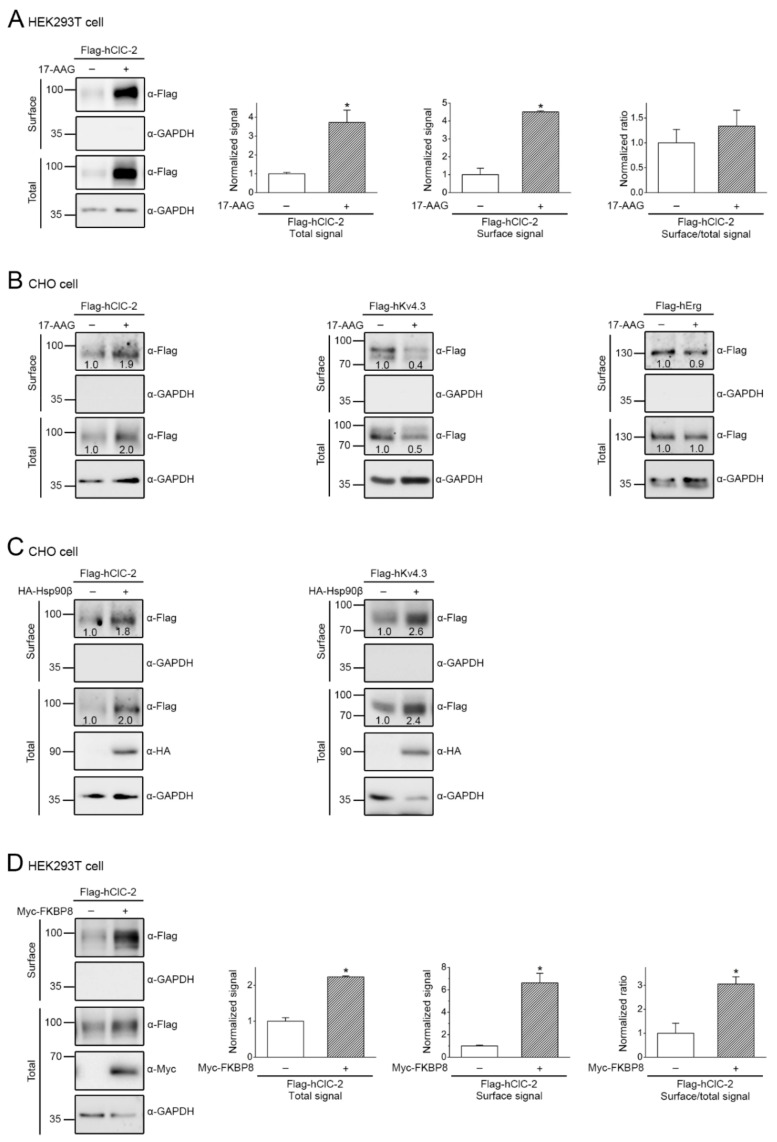
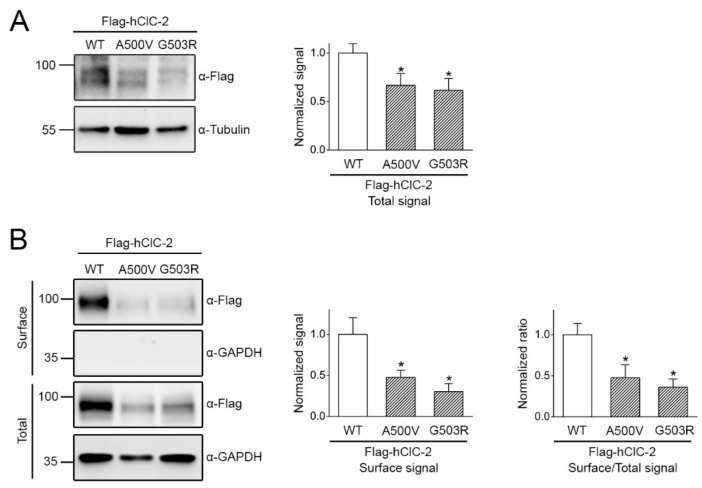
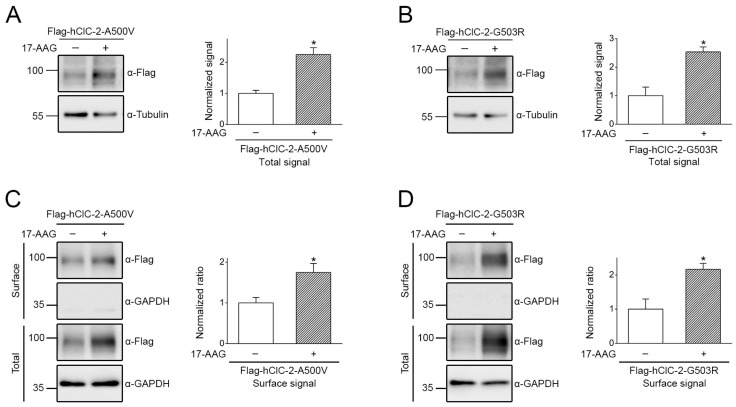
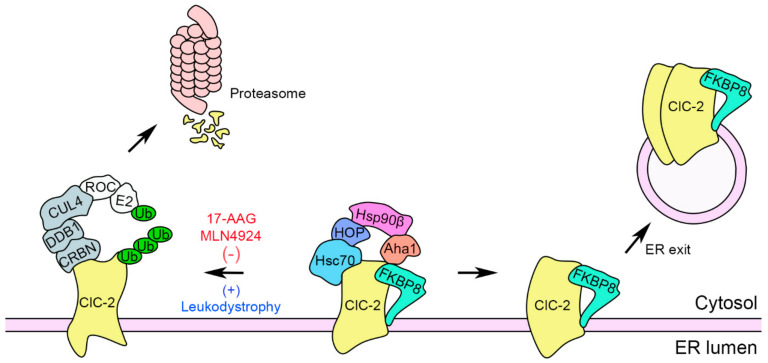
The ClC-2 channel plays a critical role in maintaining ion homeostasis in the brain and the testis. Loss-of-function mutations in the ClC-2-encoding human CLCN2 gene are linked to the white matter disease leukodystrophy. Clcn2-deficient mice display neuronal myelin vacuolation and testicular degeneration. Leukodystrophy-causing ClC-2 mutant channels are associated with anomalous proteostasis manifesting enhanced endoplasmic reticulum (ER)-associated degradation. The molecular nature of the ER quality control system for ClC-2 protein remains elusive. In mouse testicular tissues and Leydig cells, we demonstrated that endogenous ClC-2 co-existed in the same protein complex with the molecular chaperones heat shock protein 90β (Hsp90β) and heat shock cognate protein (Hsc70), as well as the associated co-chaperones Hsp70/Hsp90 organizing protein (HOP), activator of Hsp90 ATPase homolog 1 (Aha1), and FK506-binding protein 8 (FKBP8). Further biochemical analyses revealed that the Hsp90β-Hsc70 chaperone/co-chaperone system promoted mouse and human ClC-2 protein biogenesis. FKBP8 additionally facilitated membrane trafficking of ClC-2 channels. Interestingly, treatment with the Hsp90-targeting small molecule 17-allylamino-17-demethoxygeldanamycin (17-AAG) substantially boosted ClC-2 protein expression. Also, 17-AAG effectively increased both total and cell surface protein levels of leukodystrophy-causing loss-of-function ClC-2 mutant channels. Our findings highlight the therapeutic potential of 17-AAG in correcting anomalous ClC-2 proteostasis associated with leukodystrophy.
Keywords: 17-AAG; channelopathy; chaperone; co-chaperone; protein quality control; proteostasis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
