

Global Chromatin Changes Resulting from Single-Gene Inactivation-The Role of SMARCB1 in Malignant Rhabdoid Tumor
- PMID: 34071089
- PMCID: PMC8197137
- DOI: 10.3390/cancers13112561
Global Chromatin Changes Resulting from Single-Gene Inactivation-The Role of SMARCB1 in Malignant Rhabdoid Tumor
Abstract
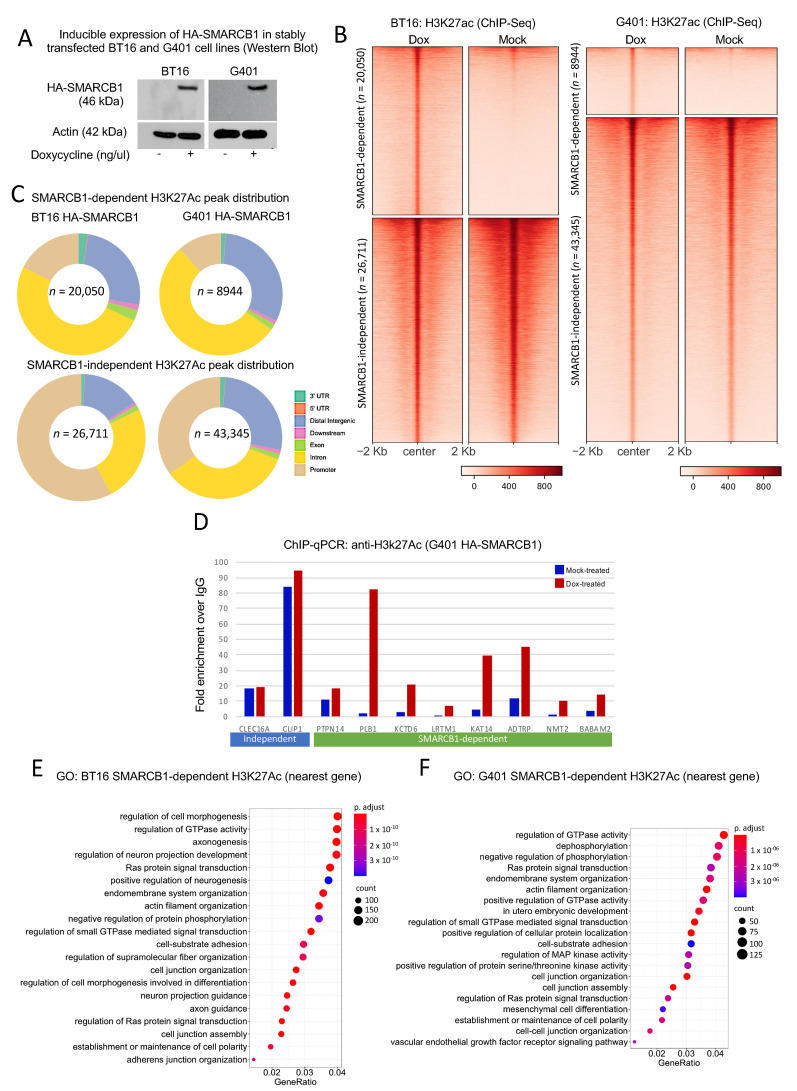
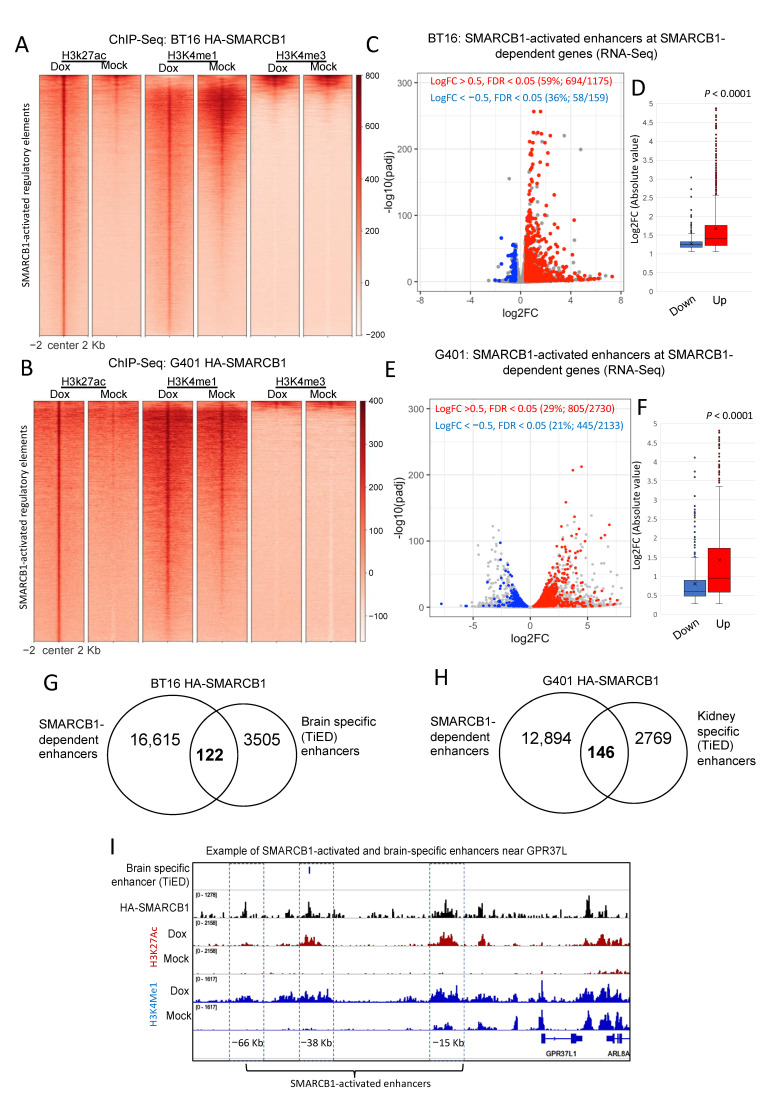
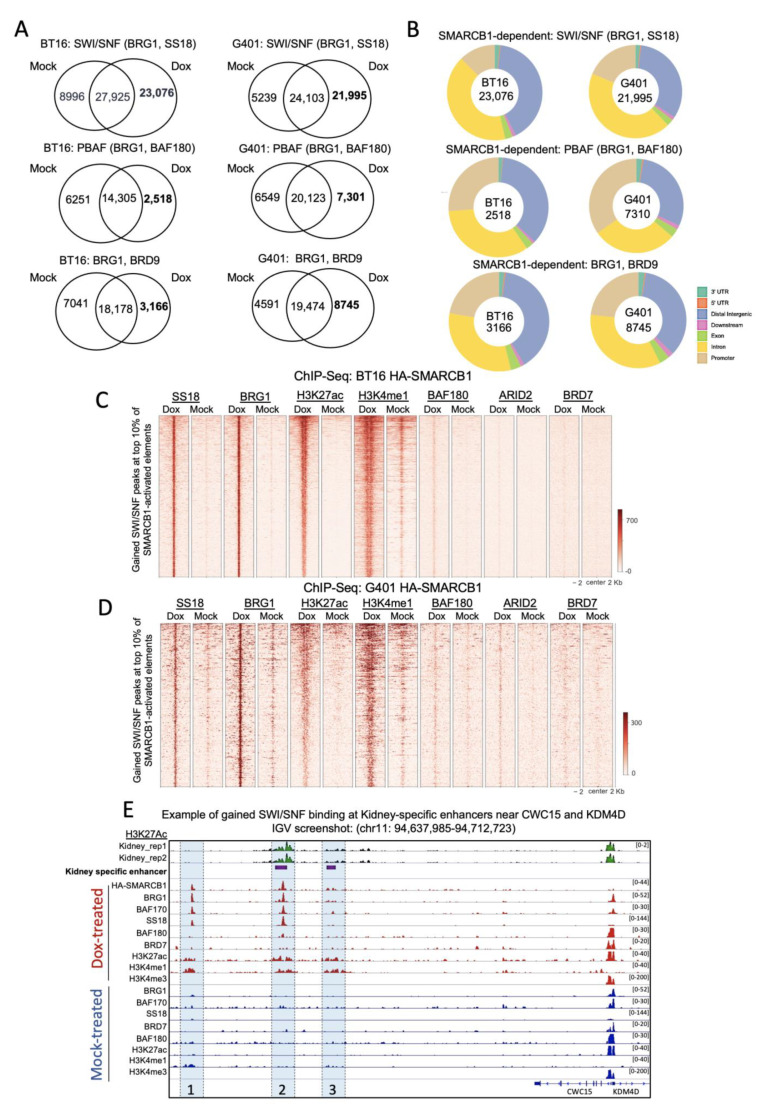
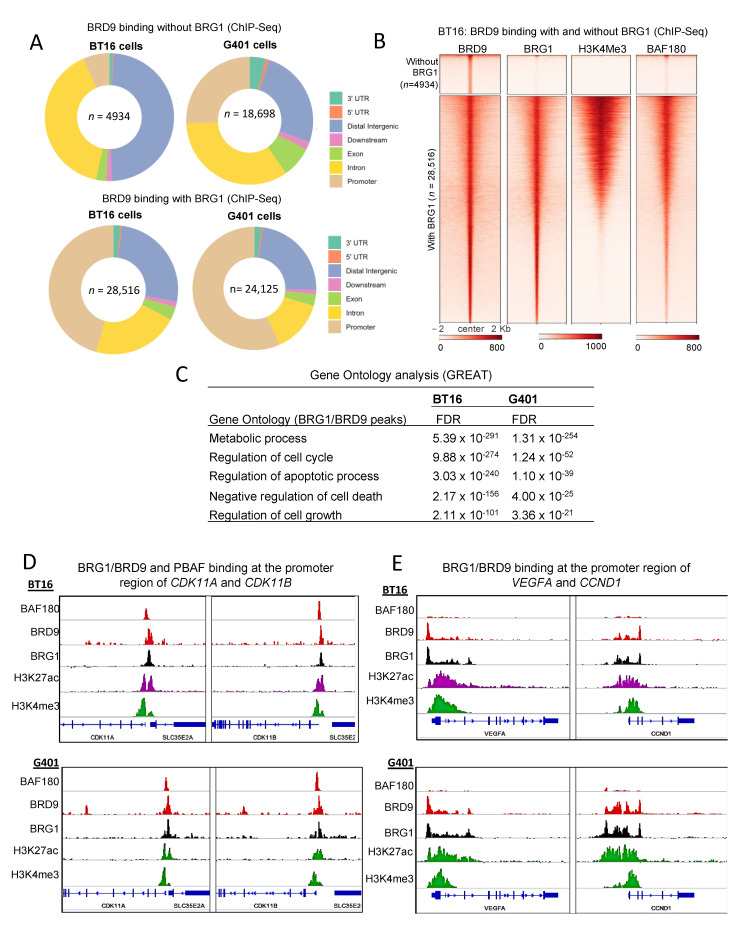
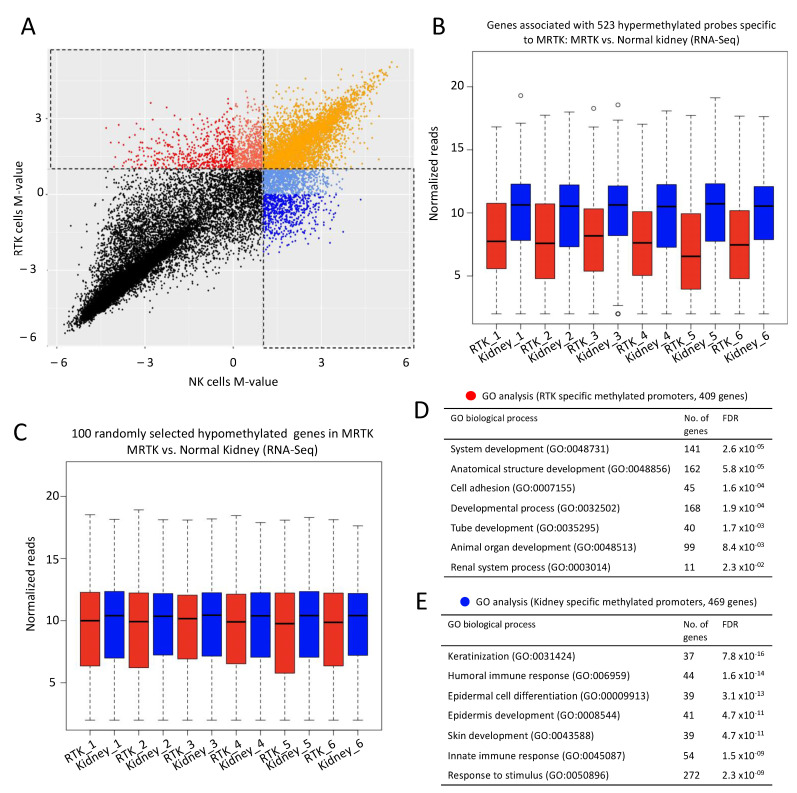
Human cancer typically results from the stochastic accumulation of multiple oncogene-activating and tumor-suppressor gene-inactivating mutations. However, this process takes time and especially in the context of certain pediatric cancer, fewer but more 'impactful' mutations may in short order produce the full-blown cancer phenotype. This is well exemplified by the highly aggressive malignant rhabdoid tumor (MRT), where the only gene classically showing recurrent inactivation is SMARCB1, a subunit member of the BAF chromatin-remodeling complex. This is true of all three presentations of MRT including MRT of kidney (MRTK), MRT of the central nervous system (atypical teratoid rhabdoid tumor-ATRT) and extracranial, extrarenal rhabdoid tumor (EERT). Our reverse modeling of rhabdoid tumors with isogenic cell lines, either induced or not induced, to express SMARCB1 showed widespread differential chromatin remodeling indicative of altered BAF complex activity with ensuant histone modifications when tested by chromatin immunoprecipitation followed by sequencing (ChIP-seq). The changes due to reintroduction of SMARCB1 were preponderantly at typical enhancers with tandem BAF complex occupancy at these sites and related gene activation, as substantiated also by transcriptomic data. Indeed, for both MRTK and ATRT cells, there is evidence of an overlap between SMARCB1-dependent enhancer activation and tissue-specific lineage-determining genes. These genes are inactive in the tumor state, conceivably arresting the cells in a primitive/undifferentiated state. This epigenetic dysregulation from inactivation of a chromatin-remodeling complex subunit contributes to an improved understanding of the complex pathophysiological basis of MRT, one of the most lethal and aggressive human cancers.
Keywords: BAF; PBAF; SMARCB1; SWI/SNF; chromatin remodeling; lineage differentiation; ncBAF; rhabdoid tumors; tissue differentiation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Biegel J.A., Zhou J.Y., Rorke L.B., Stenstrom C., Wainwright L.M., Fogelgren B. Germ-line and acquired mutations of INI1 in atyp-ical teratoid and rhabdoid tumors. Cancer Res. 1999;59:74–79. - PubMed
-
- Hasselblatt M., Gesk S., Oyen F., Rossi S., Viscardi E., Giangaspero F., Giannini C., Judkins A.R., Frühwald M.C., Obser T., et al. Nonsense Mutation and Inactivation of SMARCA4 (BRG1) in an Atypical Teratoid/Rhabdoid Tumor Showing Retained SMARCB1 (INI1) Expression. Am. J. Surg. Pathol. 2011;35:933–935. doi: 10.1097/PAS.0b013e3182196a39. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
