

Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis
- PMID: 34072544
- PMCID: PMC8198892
- DOI: 10.3390/ijms22115843
Erythrocytes: Central Actors in Multiple Scenes of Atherosclerosis
Abstract
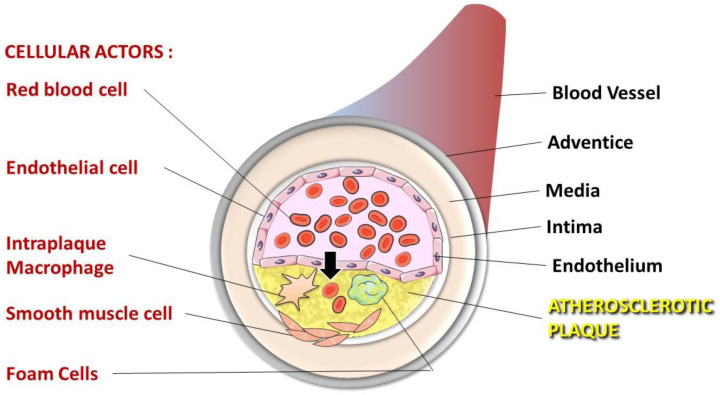
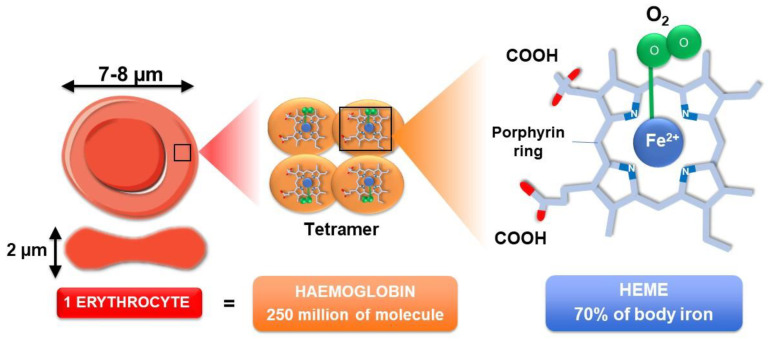
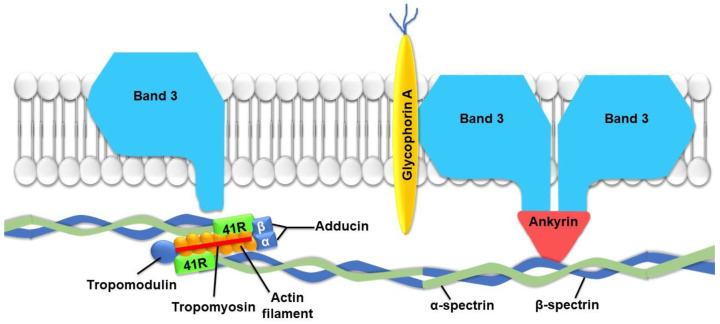
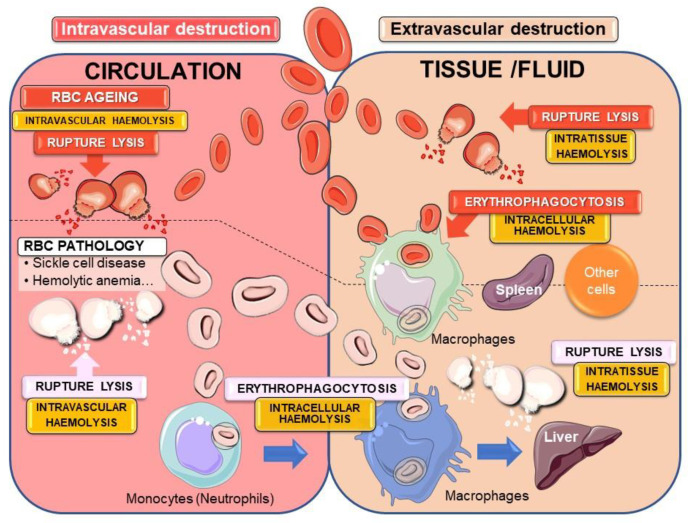
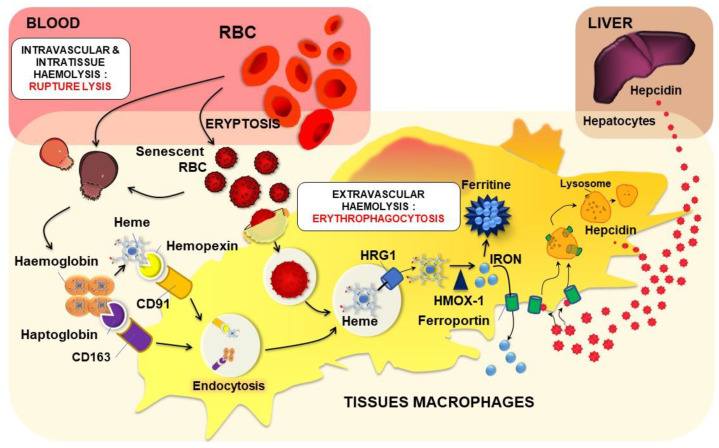
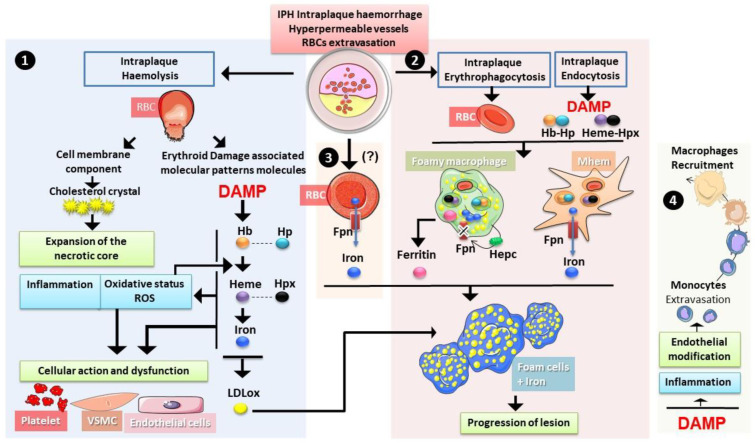
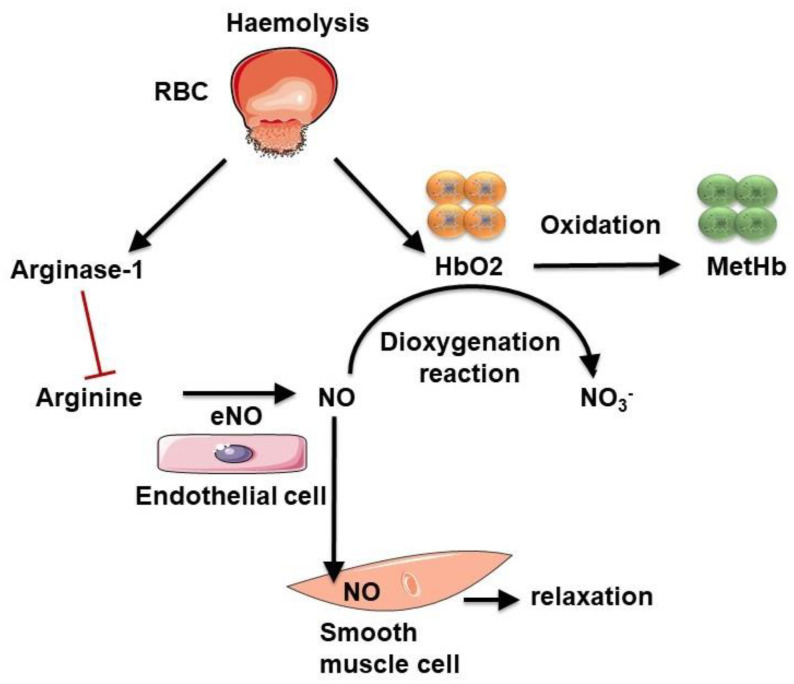
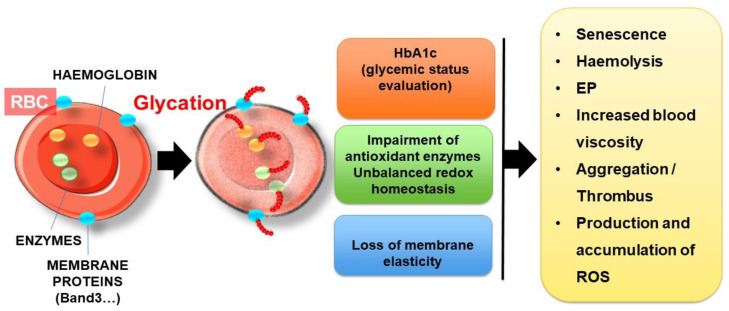
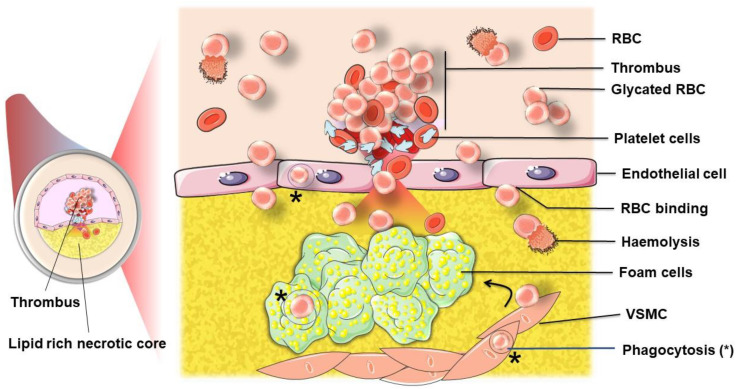
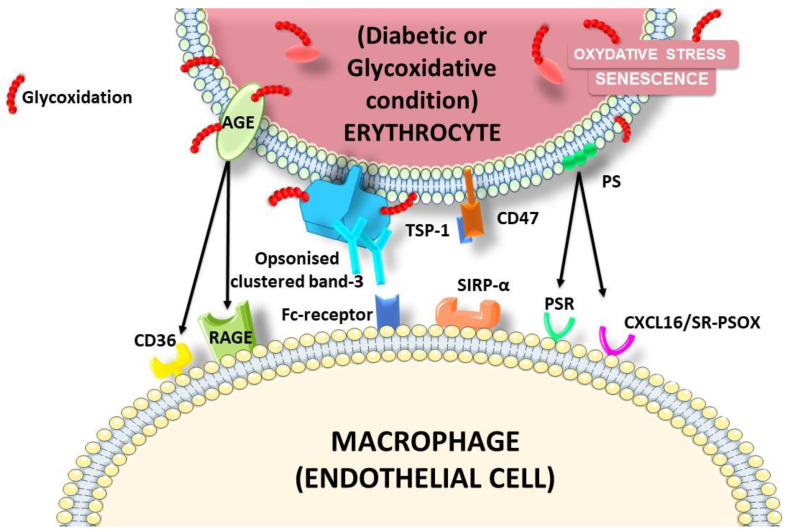
The development and progression of atherosclerosis (ATH) involves lipid accumulation, oxidative stress and both vascular and blood cell dysfunction. Erythrocytes, the main circulating cells in the body, exert determinant roles in the gas transport between tissues. Erythrocytes have long been considered as simple bystanders in cardiovascular diseases, including ATH. This review highlights recent knowledge concerning the role of erythrocytes being more than just passive gas carriers, as potent contributors to atherosclerotic plaque progression. Erythrocyte physiology and ATH pathology is first described. Then, a specific chapter delineates the numerous links between erythrocytes and atherogenesis. In particular, we discuss the impact of extravasated erythrocytes in plaque iron homeostasis with potential pathological consequences. Hyperglycaemia is recognised as a significant aggravating contributor to the development of ATH. Then, a special focus is made on glycoxidative modifications of erythrocytes and their role in ATH. This chapter includes recent data proposing glycoxidised erythrocytes as putative contributors to enhanced atherothrombosis in diabetic patients.
Keywords: atherosclerosis; eryptosis; erythrocytes; erythrophagocytosis; glycation; haemoglobin; heme; iron; oxidative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R., et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. - DOI - PubMed
-
- Palasubramaniam J., Wang X., Peter K. Myocardial Infarction-From Atherosclerosis to Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019;39:e176–e185. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
