

DHA Protects Hepatocytes from Oxidative Injury through GPR120/ERK-Mediated Mitophagy
- PMID: 34073582
- PMCID: PMC8198367
- DOI: 10.3390/ijms22115675
DHA Protects Hepatocytes from Oxidative Injury through GPR120/ERK-Mediated Mitophagy
Abstract
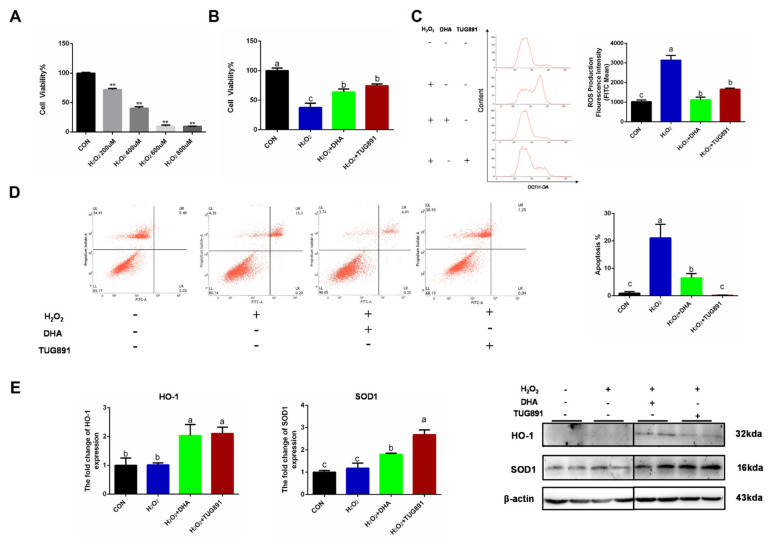
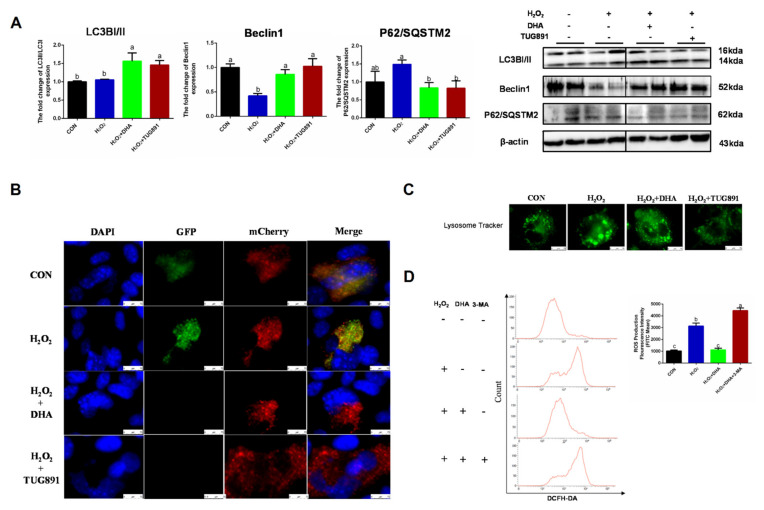
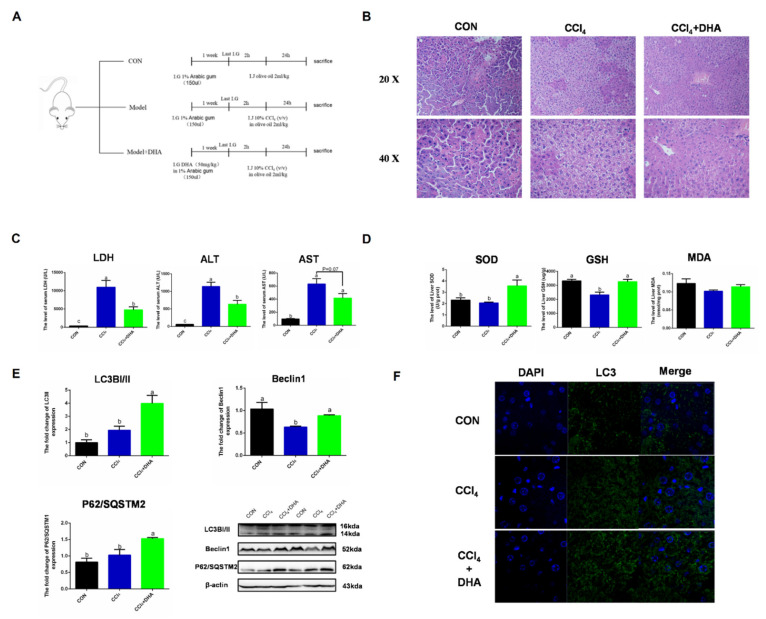
Oxidative stress occurs in a variety of clinical liver diseases and causes cellular damage and mitochondrial dysfunction. The clearance of damaged mitochondria by mitophagy may facilitate mitochondrial biogenesis and enhance cell survival. Although the supplementation of docosahexaenoic acid (DHA) has been recognized to relieve the symptoms of various liver diseases, the antioxidant effect of DHA in liver disease is still unclear. The purpose of our research was to investigate the antioxidant effect of DHA in the liver and the possible role of mitophagy in this. In vitro, H2O2-induced injury was caused in AML12 cells. The results showed that DHA repressed the level of reactive oxygen species (ROS) induced by H2O2 and stimulated the cellular antioxidation response. Most notably, DHA restored oxidative stress-impaired autophagic flux and promoted protective autophagy. In addition, PINK/Parkin-mediated mitophagy was activated by DHA in AML12 cells and alleviated mitochondrial dysfunction. The ERK1/2 signaling pathway was inhibited during oxidative stress but reactivated by DHA treatment. It was proven that the expression of ERK1/2 was involved in the regulation of mitophagy by the ERK1/2 inhibitor. We further proved these results in vivo. DHA effectively alleviated the liver oxidative damage caused by CCl4 and enhanced antioxidation capacity; intriguingly, autophagy was also activated. In summary, our data demonstrated that DHA protected hepatocytes from oxidative damage through GPR120/ERK-mediated mitophagy.
Keywords: DHA; ERK1/2 signaling; liver injury; mitophagy; oxidative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
