

SARS-CoV-2 variants, spike mutations and immune escape
- PMID: 34075212
- PMCID: PMC8167834
- DOI: 10.1038/s41579-021-00573-0
SARS-CoV-2 variants, spike mutations and immune escape
Abstract
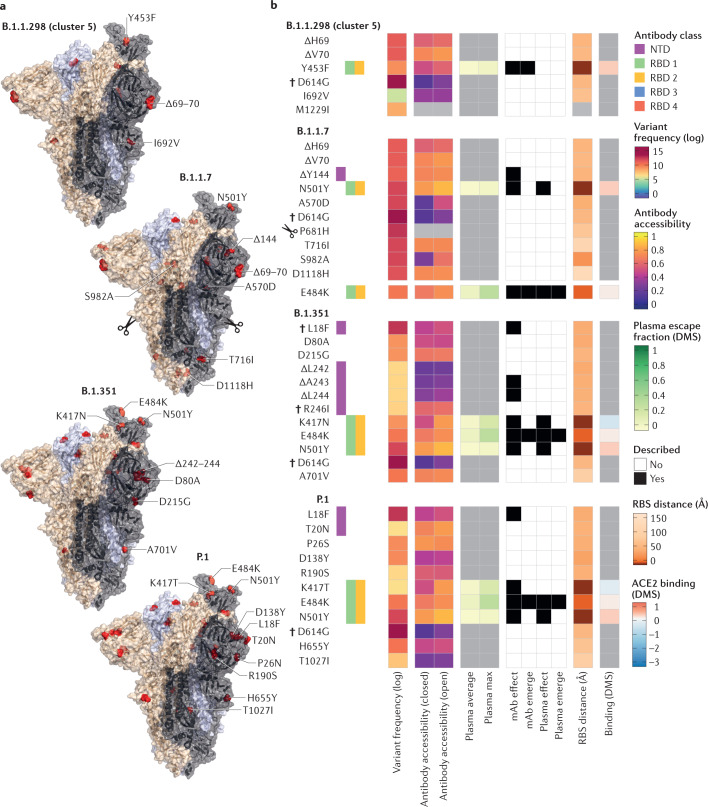
Although most mutations in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome are expected to be either deleterious and swiftly purged or relatively neutral, a small proportion will affect functional properties and may alter infectivity, disease severity or interactions with host immunity. The emergence of SARS-CoV-2 in late 2019 was followed by a period of relative evolutionary stasis lasting about 11 months. Since late 2020, however, SARS-CoV-2 evolution has been characterized by the emergence of sets of mutations, in the context of 'variants of concern', that impact virus characteristics, including transmissibility and antigenicity, probably in response to the changing immune profile of the human population. There is emerging evidence of reduced neutralization of some SARS-CoV-2 variants by postvaccination serum; however, a greater understanding of correlates of protection is required to evaluate how this may impact vaccine effectiveness. Nonetheless, manufacturers are preparing platforms for a possible update of vaccine sequences, and it is crucial that surveillance of genetic and antigenic changes in the global virus population is done alongside experiments to elucidate the phenotypic impacts of mutations. In this Review, we summarize the literature on mutations of the SARS-CoV-2 spike protein, the primary antigen, focusing on their impacts on antigenicity and contextualizing them in the protein structure, and discuss them in the context of observed mutation frequencies in global sequence datasets.
Conflict of interest statement
The authors declare no competing interests.
Figures
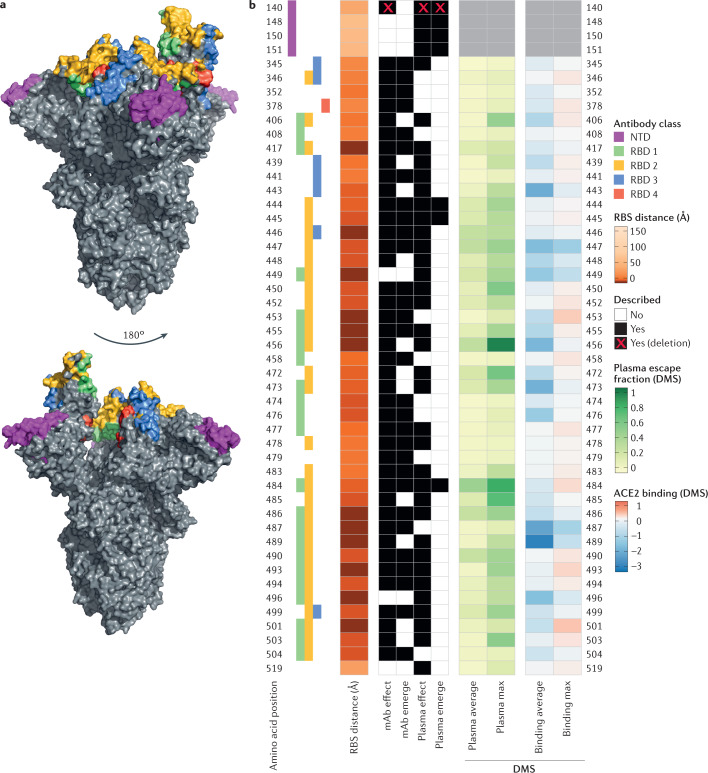
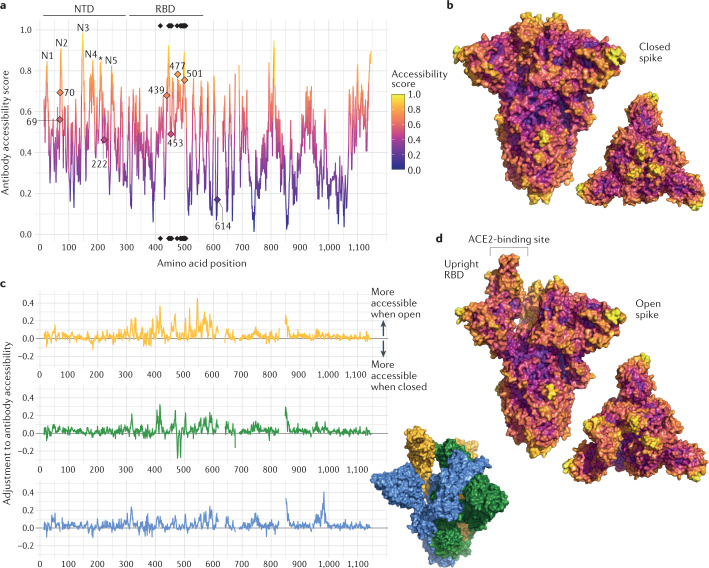
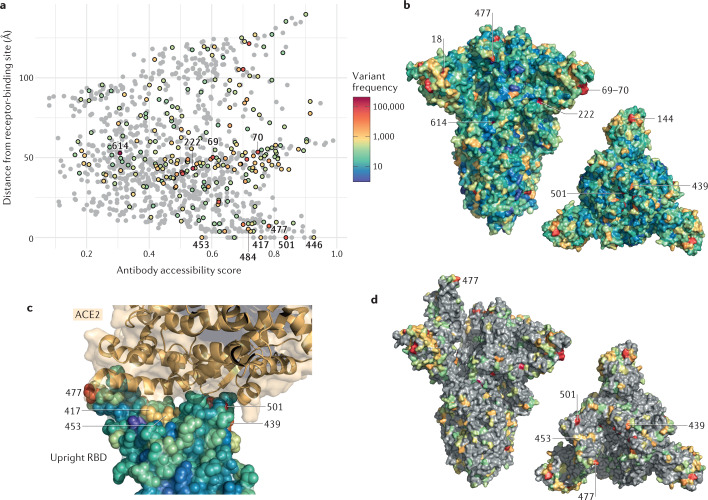
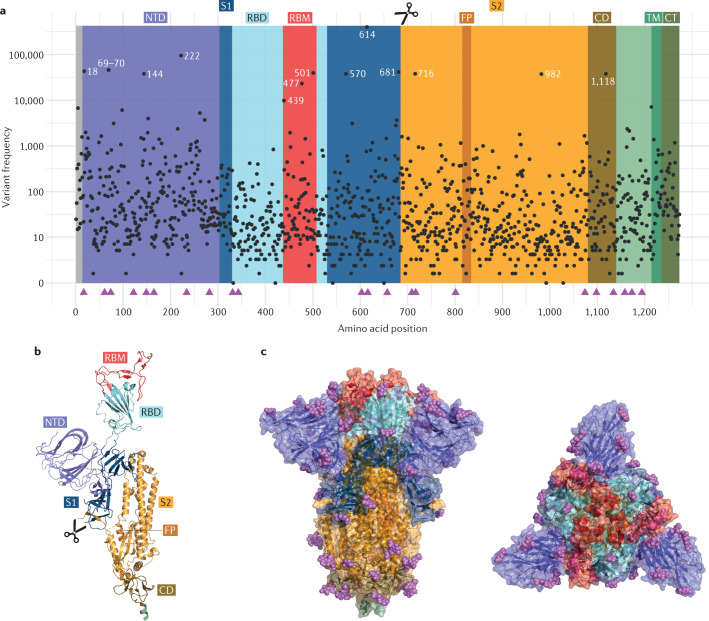
References
-
- WHO. Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (2021).
-
- Rambaut, A. et al. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel set of Spike Mutations. https://virological.org/t/preliminary-genomic-characterisation-of-an-eme... (2020).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
