

Nitric oxide signalling in kidney regulation and cardiometabolic health
- PMID: 34075241
- PMCID: PMC8169406
- DOI: 10.1038/s41581-021-00429-z
Nitric oxide signalling in kidney regulation and cardiometabolic health
Abstract
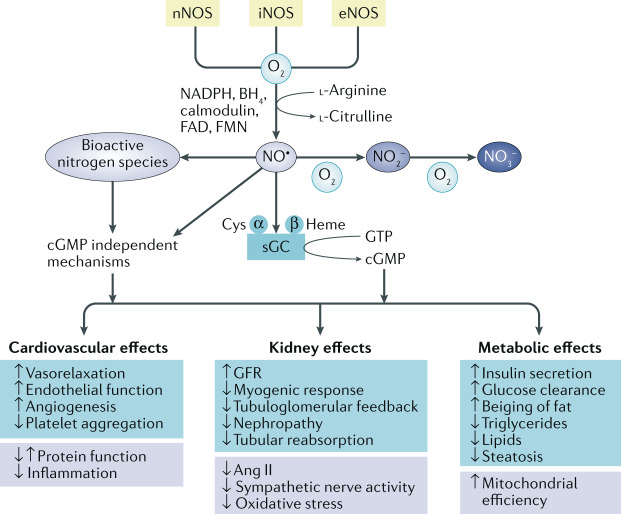
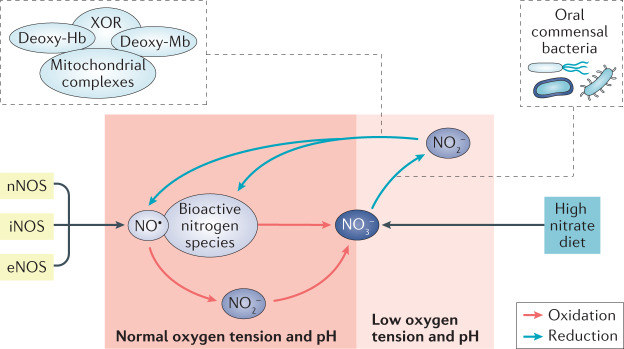
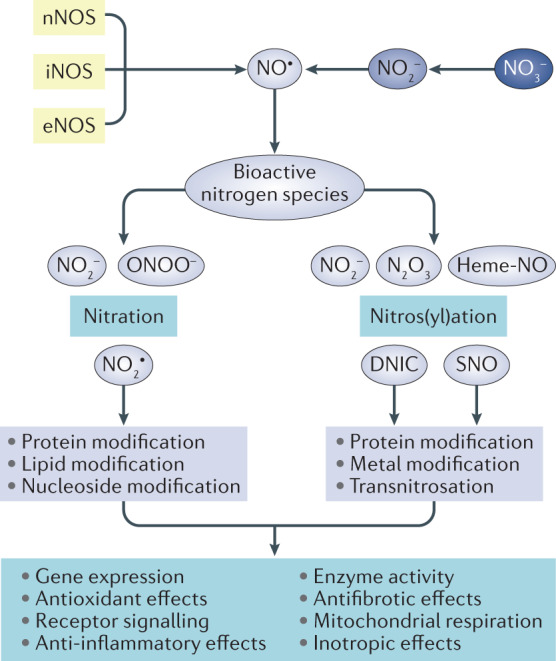
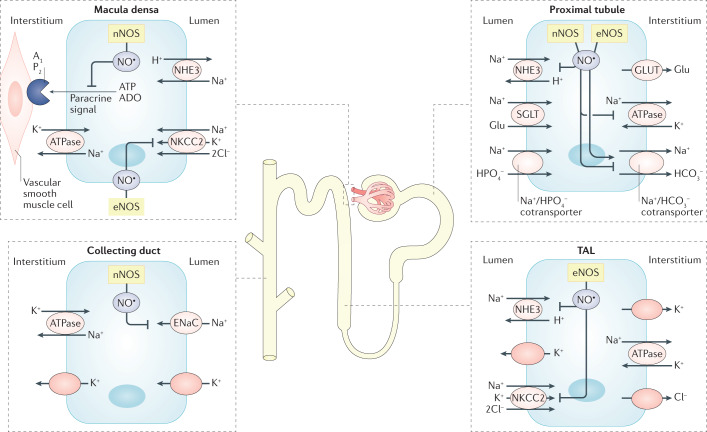
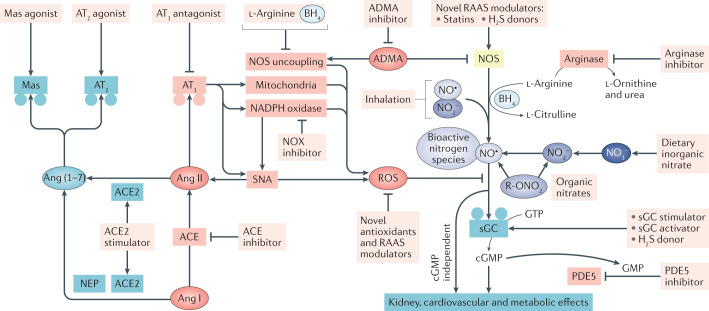
The prevalence of cardiovascular and metabolic disease coupled with kidney dysfunction is increasing worldwide. This triad of disorders is associated with considerable morbidity and mortality as well as a substantial economic burden. Further understanding of the underlying pathophysiological mechanisms is important to develop novel preventive or therapeutic approaches. Among the proposed mechanisms, compromised nitric oxide (NO) bioactivity associated with oxidative stress is considered to be important. NO is a short-lived diatomic signalling molecule that exerts numerous effects on the kidneys, heart and vasculature as well as on peripheral metabolically active organs. The enzymatic L-arginine-dependent NO synthase (NOS) pathway is classically viewed as the main source of endogenous NO formation. However, the function of the NOS system is often compromised in various pathologies including kidney, cardiovascular and metabolic diseases. An alternative pathway, the nitrate-nitrite-NO pathway, enables endogenous or dietary-derived inorganic nitrate and nitrite to be recycled via serial reduction to form bioactive nitrogen species, including NO, independent of the NOS system. Signalling via these nitrogen species is linked with cGMP-dependent and independent mechanisms. Novel approaches to restoring NO homeostasis during NOS deficiency and oxidative stress have potential therapeutic applications in kidney, cardiovascular and metabolic disorders.
© 2021. Springer Nature Limited.
Conflict of interest statement
The author declares no competing interests
Figures
References
-
- Global Burden of Metabolic Risk Factors for Chronic Diseases Collaboration. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. 2014;2:634–647. doi: 10.1016/S2213-8587(14)70102-0. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
