

Epigenomic landscape and 3D genome structure in pediatric high-grade glioma
- PMID: 34078608
- PMCID: PMC10166578
- DOI: 10.1126/sciadv.abg4126
Epigenomic landscape and 3D genome structure in pediatric high-grade glioma
Abstract
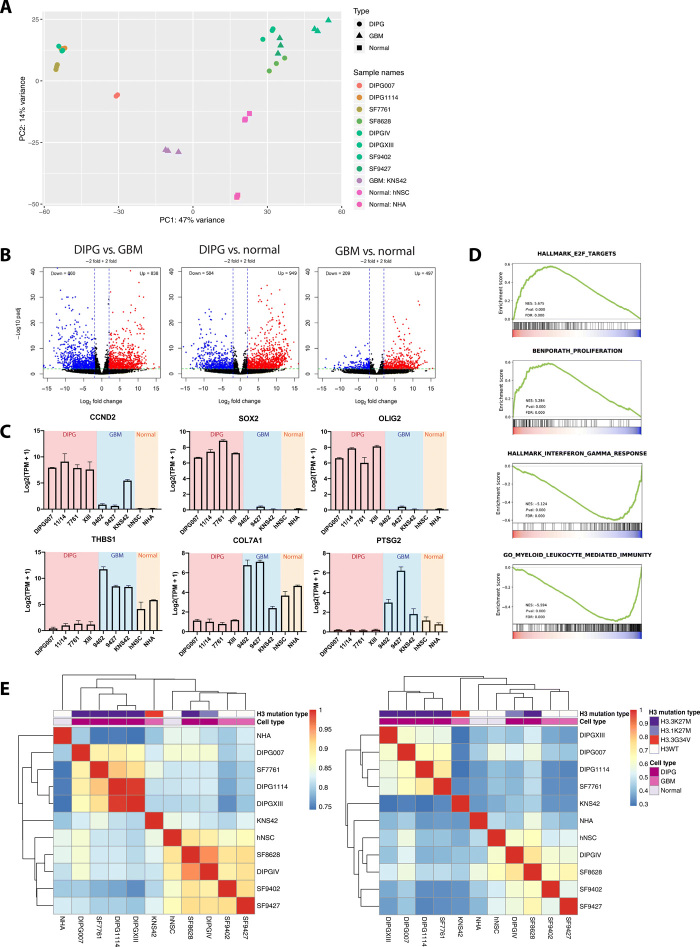
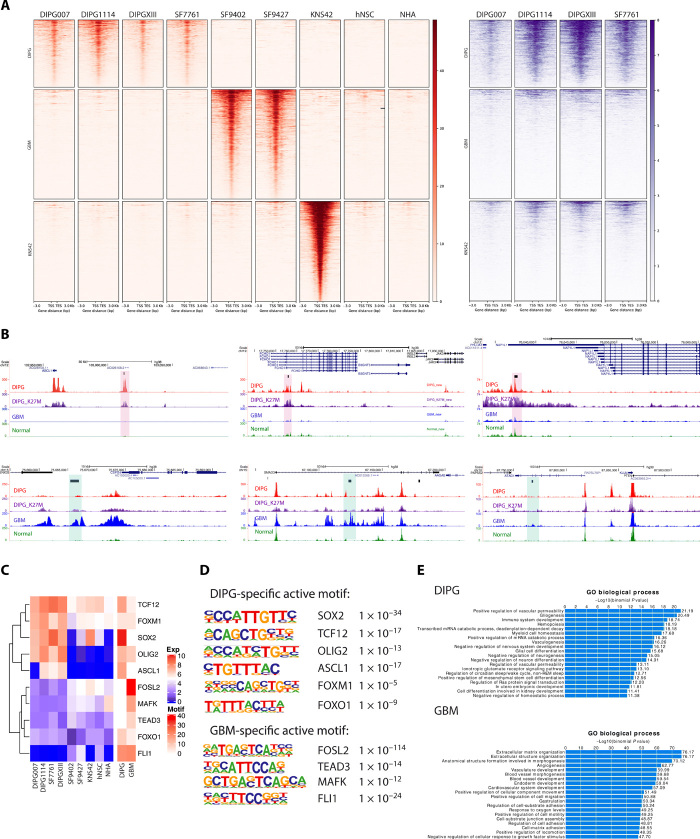
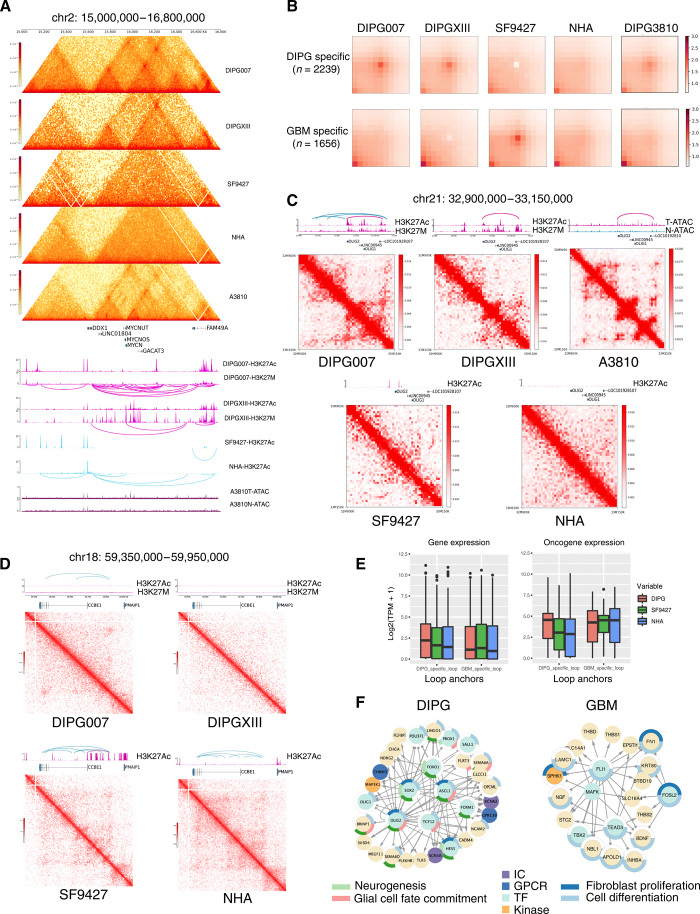
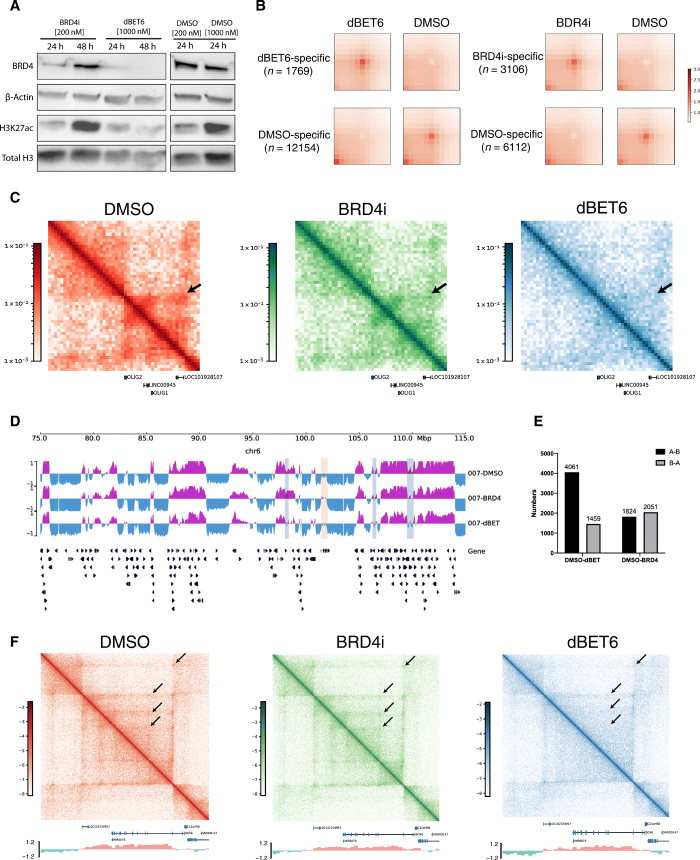
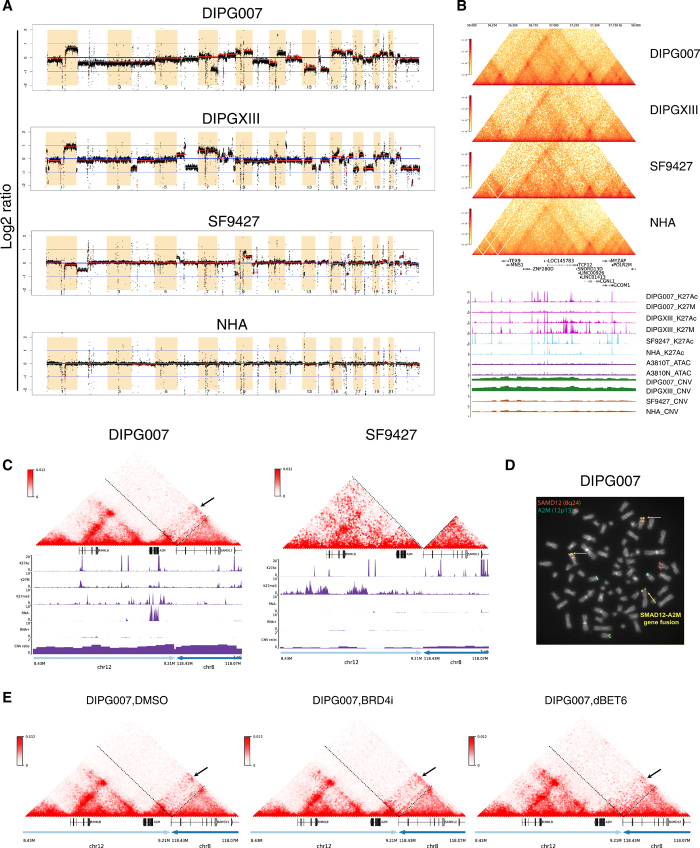
Pediatric high-grade gliomas (pHGGs), including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG), are morbid brain tumors. Even with treatment survival is poor, making pHGG the number one cause of cancer death in children. Up to 80% of DIPGs harbor a somatic missense mutation in genes encoding histone H3. To investigate whether H3K27M is associated with distinct chromatin structure that alters transcription regulation, we generated the first high-resolution Hi-C maps of pHGG cell lines and tumor tissue. By integrating transcriptome (RNA-seq), enhancer landscape (ChIP-seq), genome structure (Hi-C), and chromatin accessibility (ATAC-seq) datasets from H3K27M and wild-type specimens, we identified tumor-specific enhancers and regulatory networks for known oncogenes. We identified genomic structural variations that lead to potential enhancer hijacking and gene coamplification, including A2M, JAG2, and FLRT1 Together, our results imply three-dimensional genome alterations may play a critical role in the pHGG epigenetic landscape and contribute to tumorigenesis.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Mackay A., Burford A., Carvalho D., Izquierdo E., Fazal-Salom J., Taylor K. R., Bjerke L., Clarke M., Vinci M., Nandhabalan M., Temelso S., Popov S., Molinari V., Raman P., Waanders A. J., Han H. J., Gupta S., Marshall L., Zacharoulis S., Vaidya S., Mandeville H. C., Bridges L. R., Martin A. J., Al-Sarraj S., Chandler C., Ng H.-K., Li X., Mu K., Trabelsi S., Brahim D. H.-B., Kisljakov A. N., Konovalov D. M., Moore A. S., Carcaboso A. M., Sunol M., de Torres C., Cruz O., Mora J., Shats L. I., Stavale J. N., Bidinotto L. T., Reis R. M., Entz-Werle N., Farrell M., Cryan J., Crimmins D., Caird J., Pears J., Monje M., Debily M.-A., Castel D., Grill J., Hawkins C., Nikbakht H., Jabado N., Baker S. J., Pfister S. M., Jones D. T. W., Fouladi M., von Bueren A. O., Baudis M., Resnick A., Jones C., Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017). - PMC - PubMed
-
- Jones C., Karajannis M. A., Jones D. T. W., Kieran M. W., Monje M., Baker S. J., Becher O. J., Cho Y.-J., Gupta N., Hawkins C., Hargrave D., Haas-Kogan D. A., Jabado N., Li X.-N., Mueller S., Nicolaides T., Packer R. J., Persson A. I., Phillips J. J., Simonds E. F., Stafford J. M., Tang Y., Pfister S. M., Weiss W. A., Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro Oncol. 19, 153–161 (2017). - PMC - PubMed
-
- Nagaraja S., Quezada M. A., Gillespie S. M., Arzt M., Lennon J. J., Woo P. J., Hovestadt V., Kambhampati M., Filbin M. G., Suva M. L., Nazarian J., Monje M., Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol. Cell 76, 965–980.e12 (2019). - PMC - PubMed
-
- Stafford J. M., Lee C.-H., Voigt P., Descostes N., Saldaña-Meyer R., Yu J.-R., Leroy G., Oksuz O., Chapman J. R., Suarez F., Modrek A. S., Bayin N. S., Placantonakis D. G., Karajannis M. A., Snuderl M., Ueberheide B., Reinberg D., Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 4, eaau5935 (2018). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
