

Discovery of widespread transcription initiation at microsatellites predictable by sequence-based deep neural network
- PMID: 34078885
- PMCID: PMC8172540
- DOI: 10.1038/s41467-021-23143-7
Discovery of widespread transcription initiation at microsatellites predictable by sequence-based deep neural network
Erratum in
-
Author Correction: Discovery of widespread transcription initiation at microsatellites predictable by sequence-based deep neural network.Nat Commun. 2022 Mar 1;13(1):1200. doi: 10.1038/s41467-022-28758-y. Nat Commun. 2022. PMID: 35232988 Free PMC article. No abstract available.
Abstract
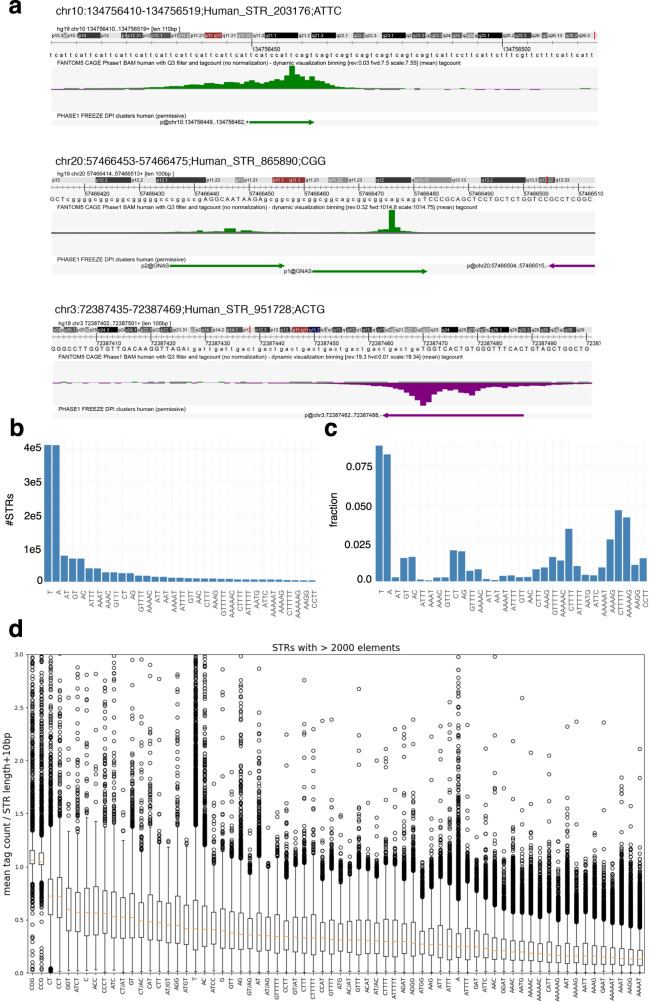
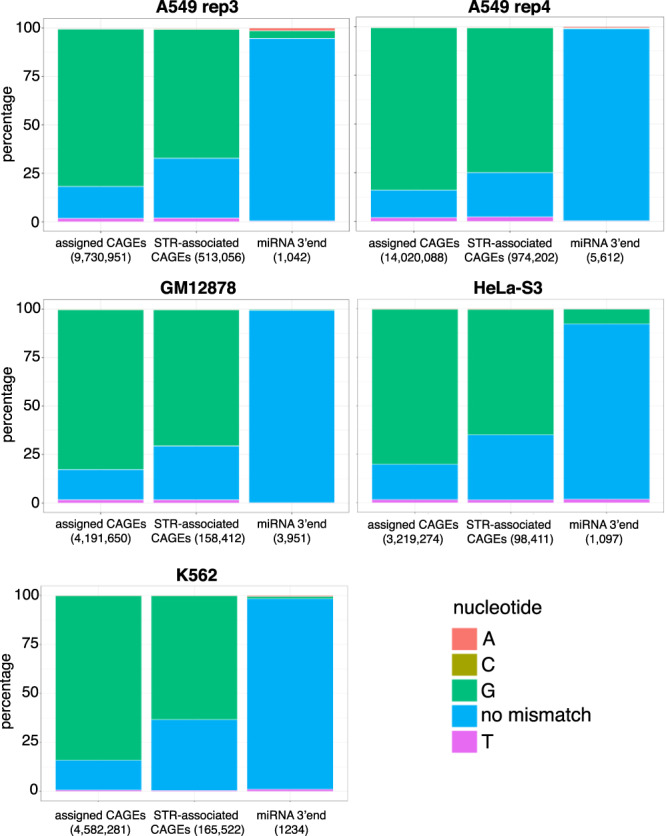
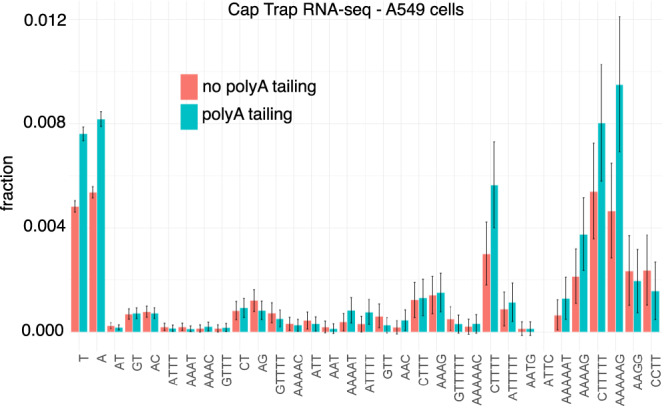
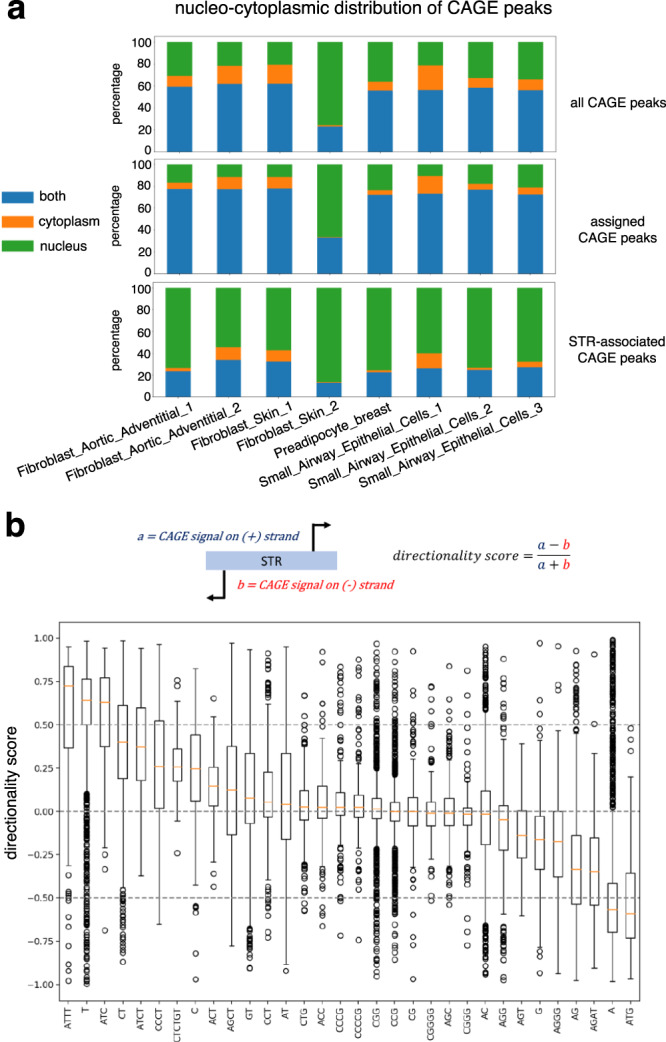
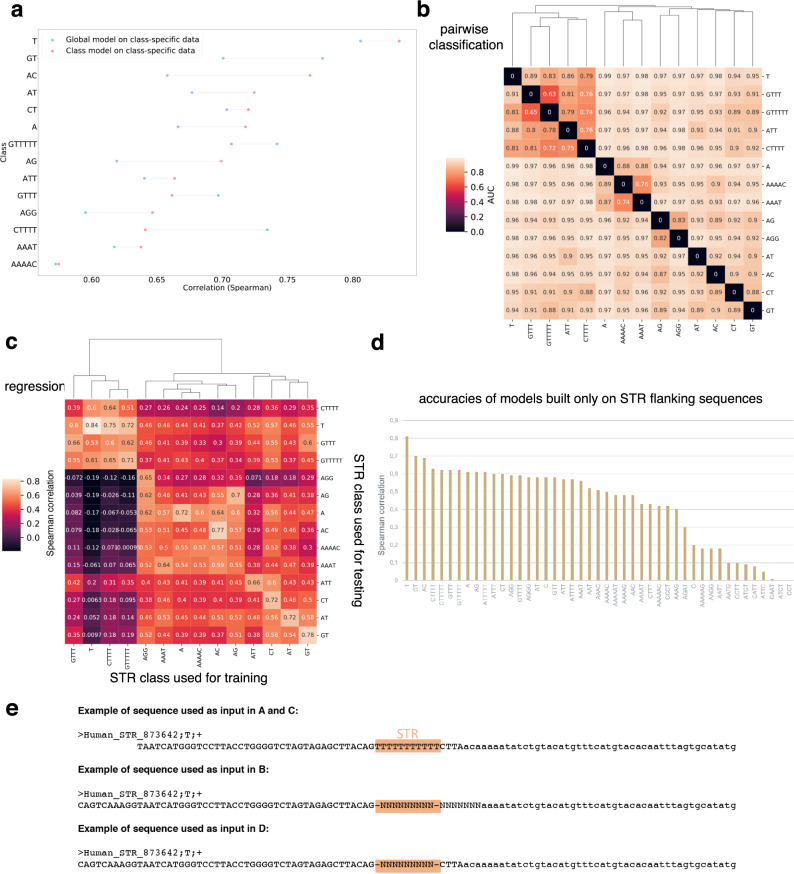
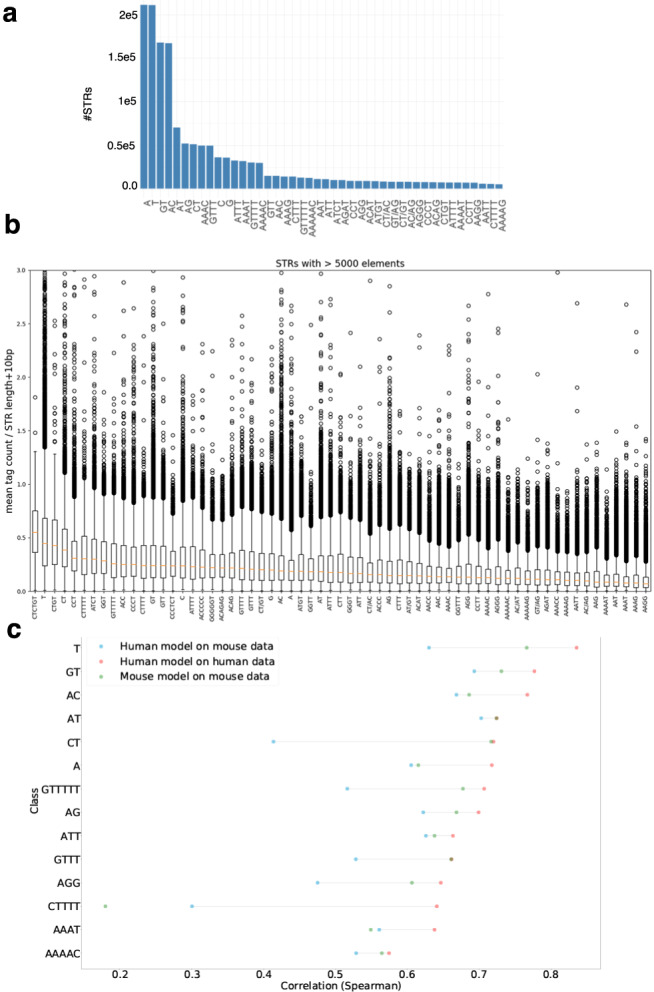
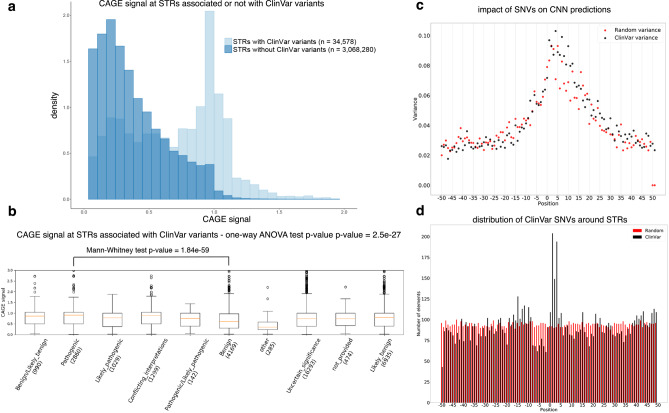
Using the Cap Analysis of Gene Expression (CAGE) technology, the FANTOM5 consortium provided one of the most comprehensive maps of transcription start sites (TSSs) in several species. Strikingly, ~72% of them could not be assigned to a specific gene and initiate at unconventional regions, outside promoters or enhancers. Here, we probe these unassigned TSSs and show that, in all species studied, a significant fraction of CAGE peaks initiate at microsatellites, also called short tandem repeats (STRs). To confirm this transcription, we develop Cap Trap RNA-seq, a technology which combines cap trapping and long read MinION sequencing. We train sequence-based deep learning models able to predict CAGE signal at STRs with high accuracy. These models unveil the importance of STR surrounding sequences not only to distinguish STR classes, but also to predict the level of transcription initiation. Importantly, genetic variants linked to human diseases are preferentially found at STRs with high transcription initiation level, supporting the biological and clinical relevance of transcription initiation at STRs. Together, our results extend the repertoire of non-coding transcription associated with DNA tandem repeats and complexify STR polymorphism.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
