

The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome
- PMID: 34079956
- PMCID: PMC8168619
- DOI: 10.1038/s43018-020-00131-2
The proliferative history shapes the DNA methylome of B-cell tumors and predicts clinical outcome
Abstract
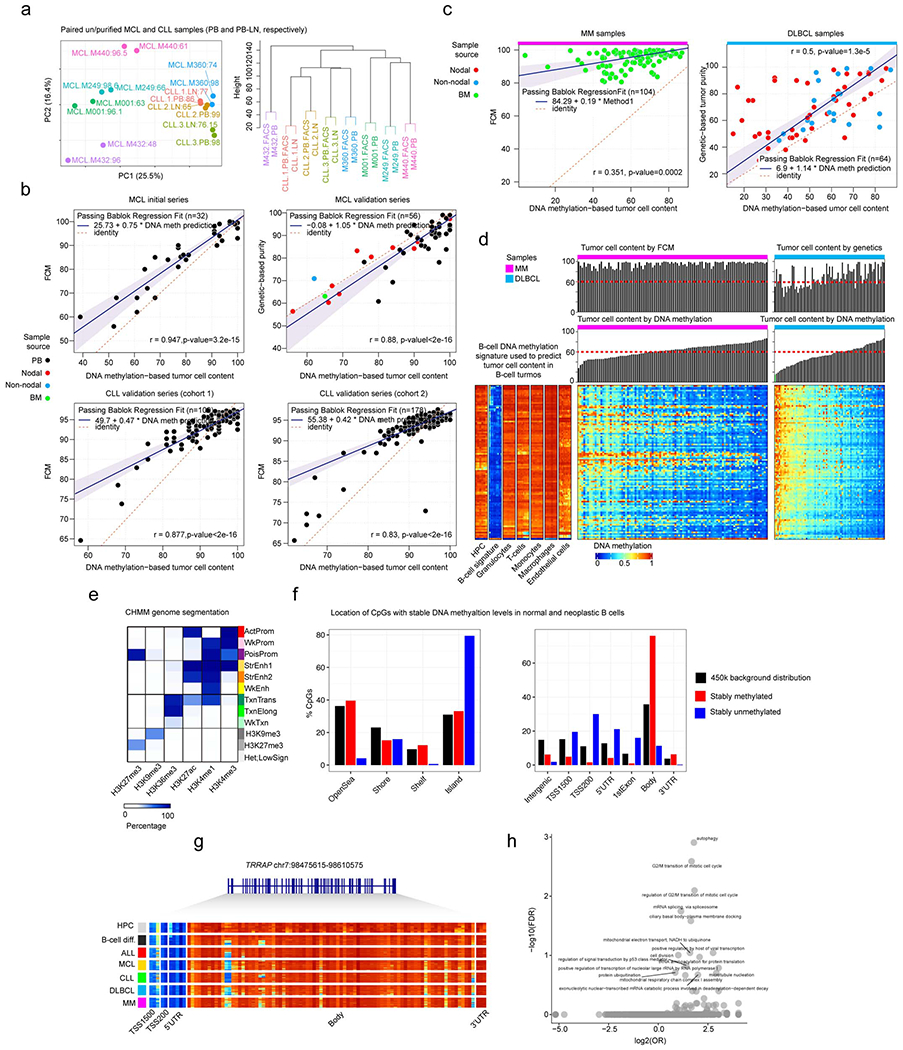
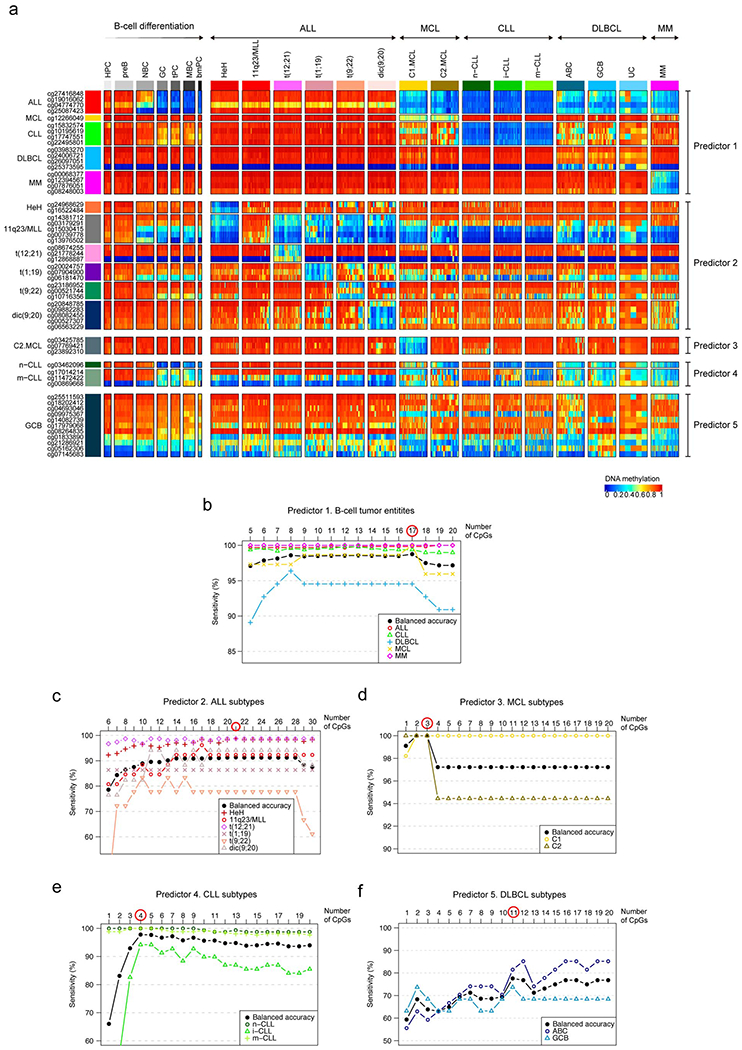
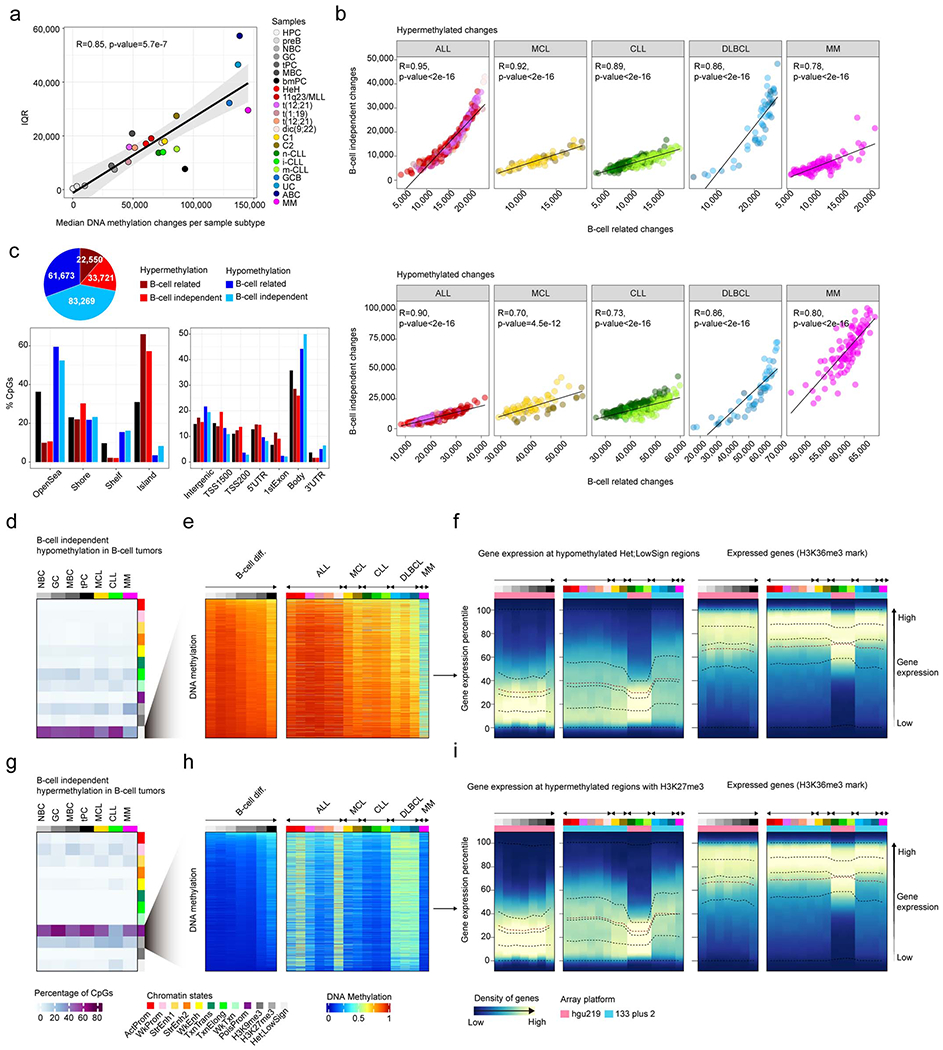
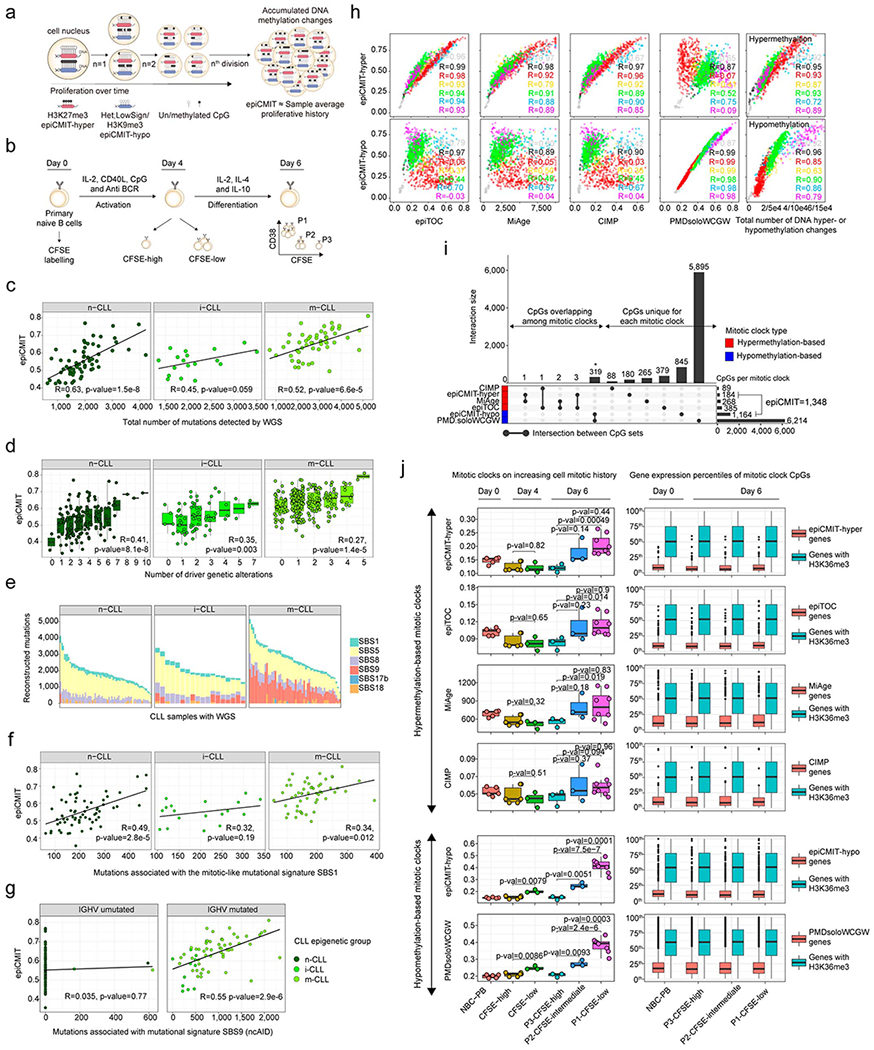
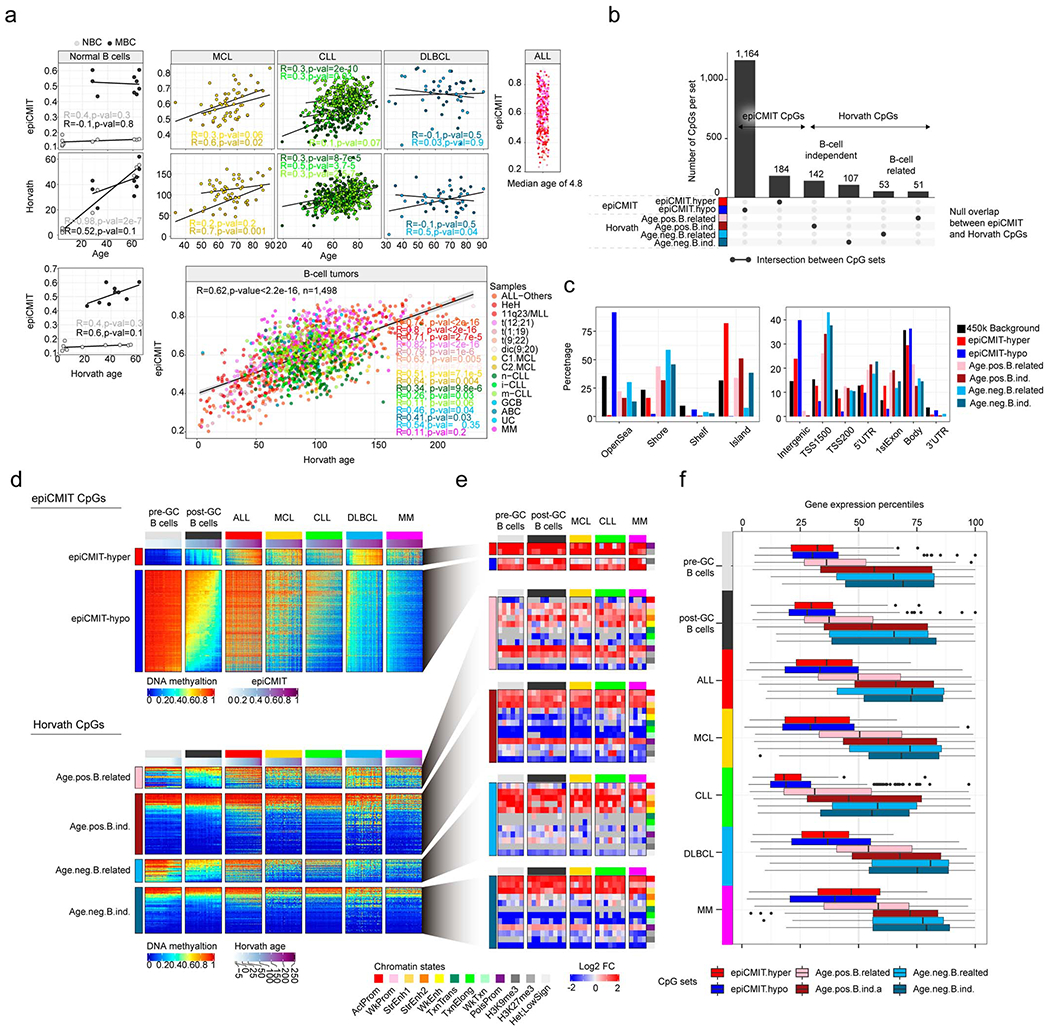
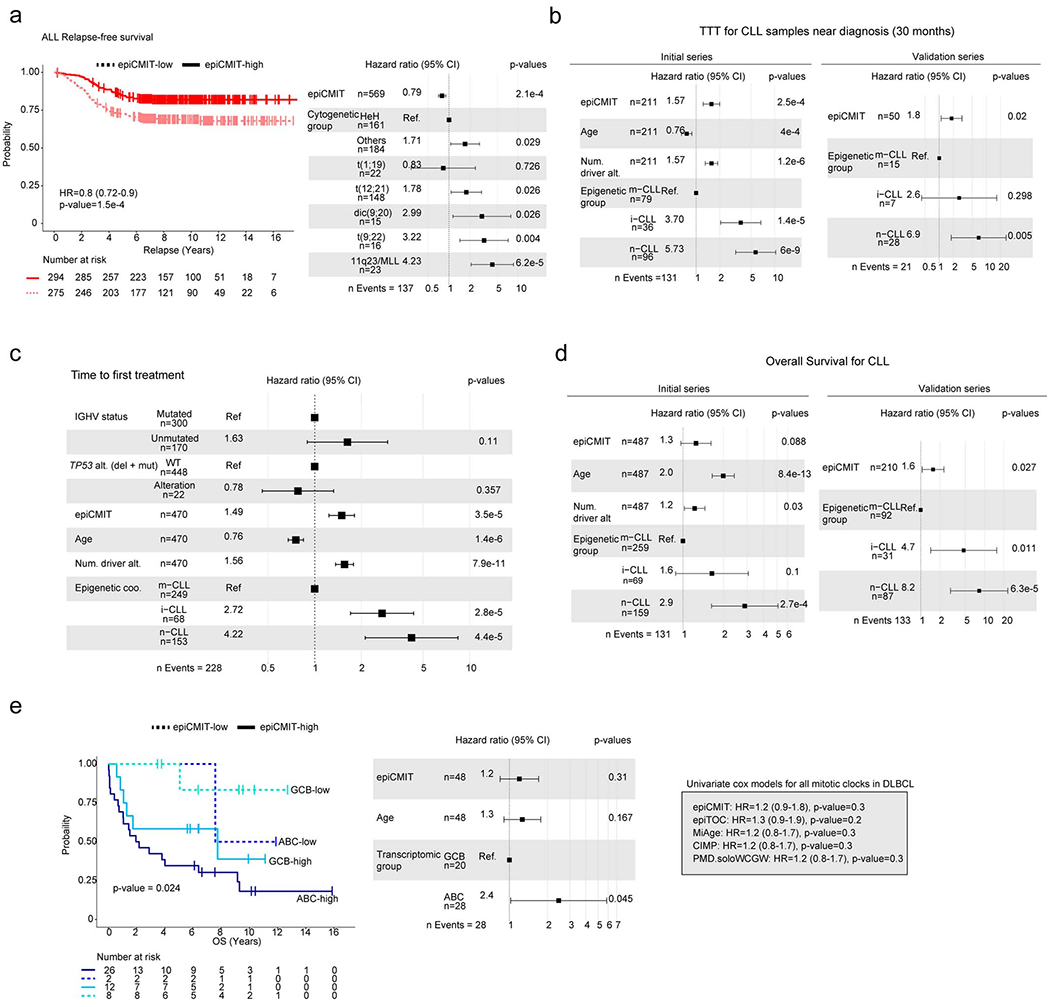
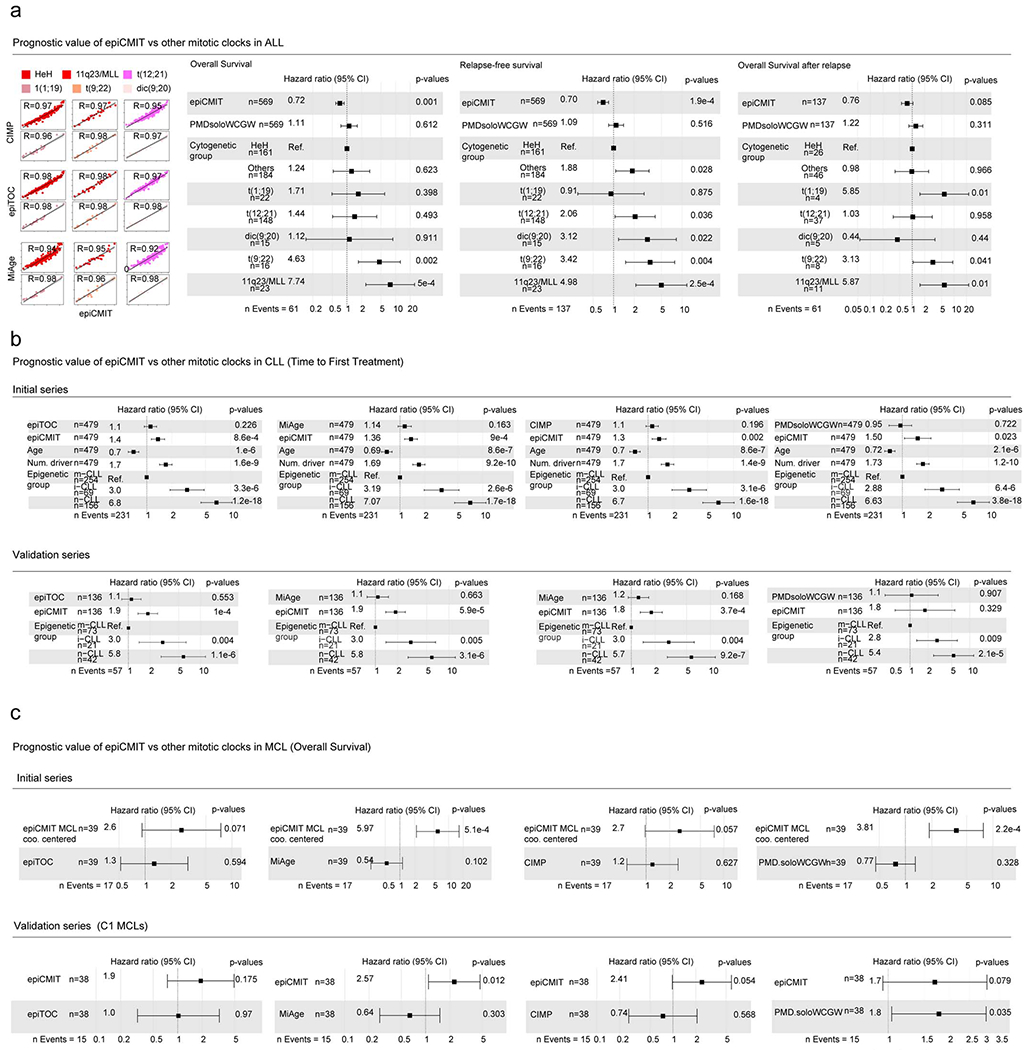
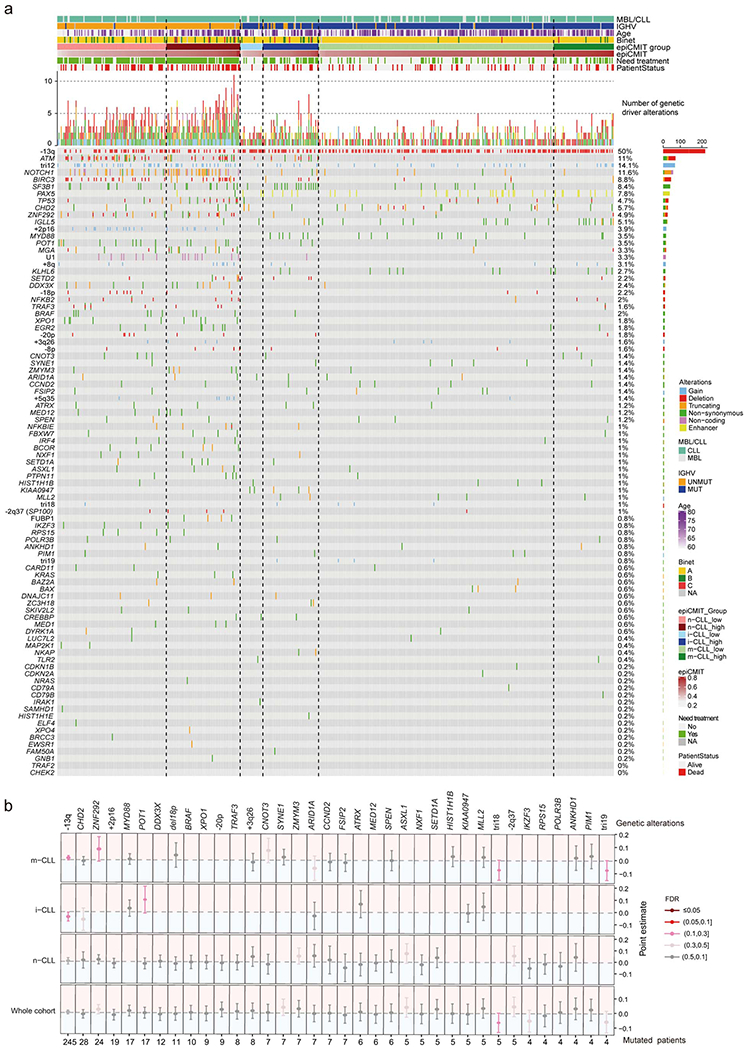
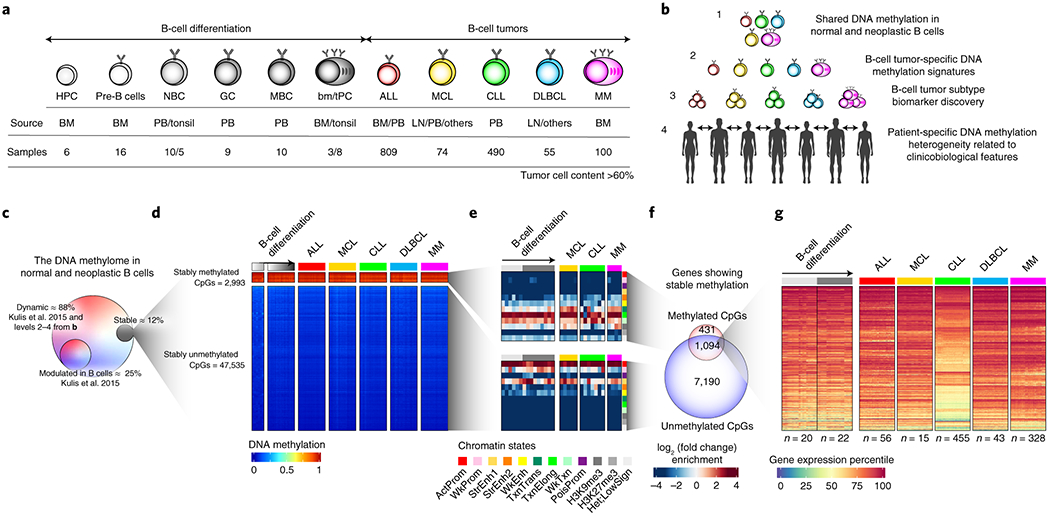
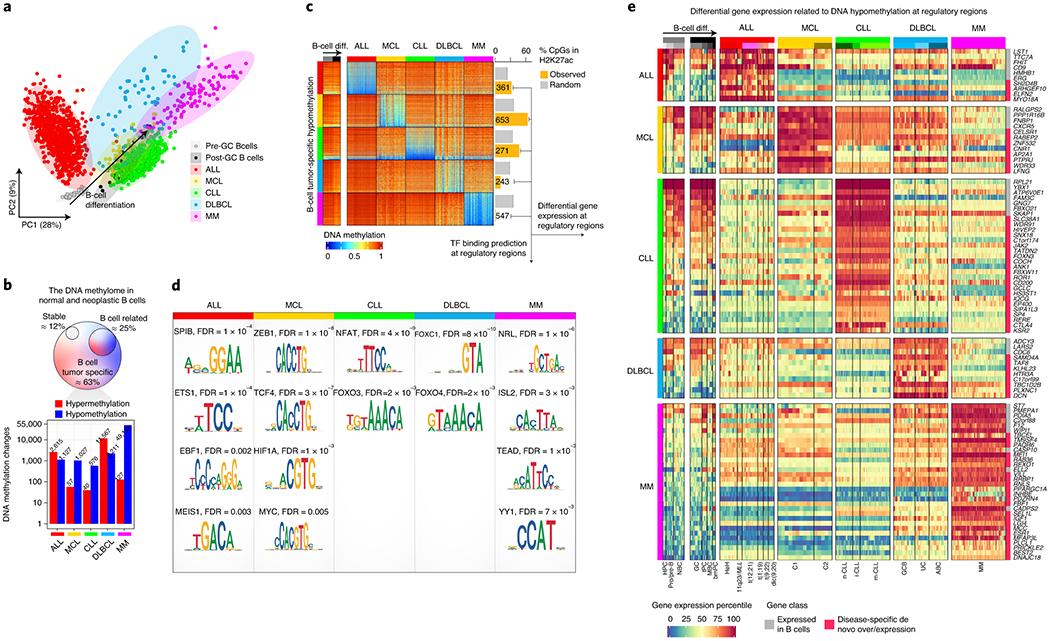
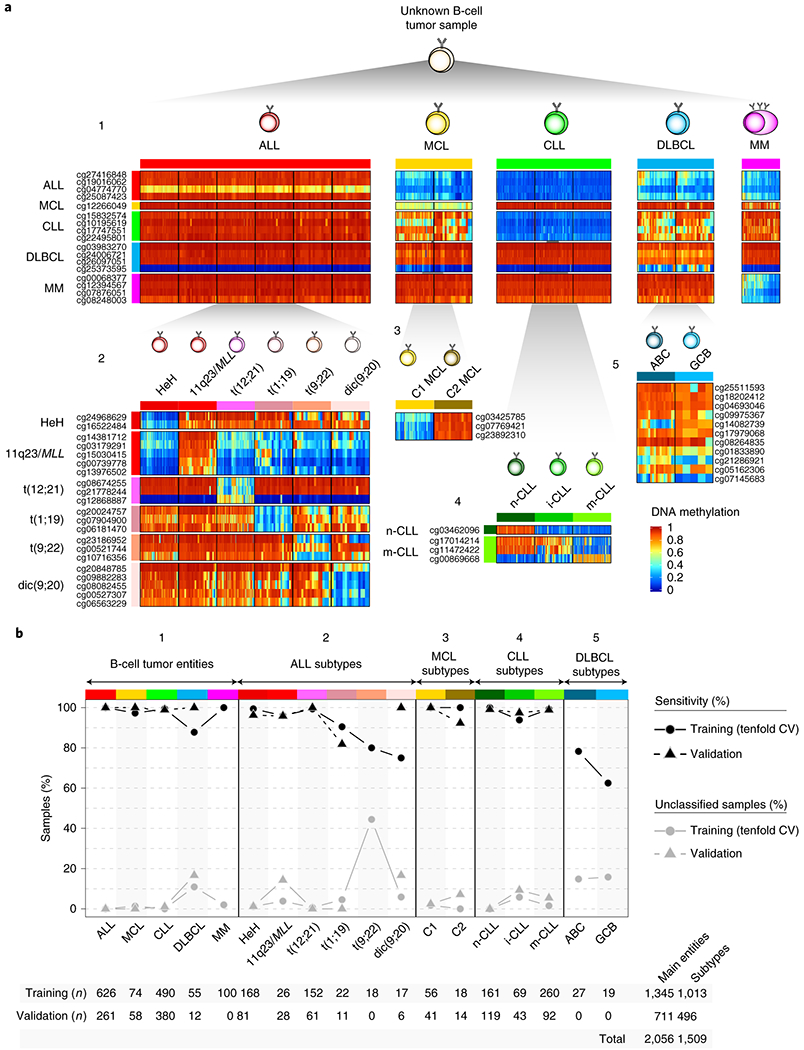
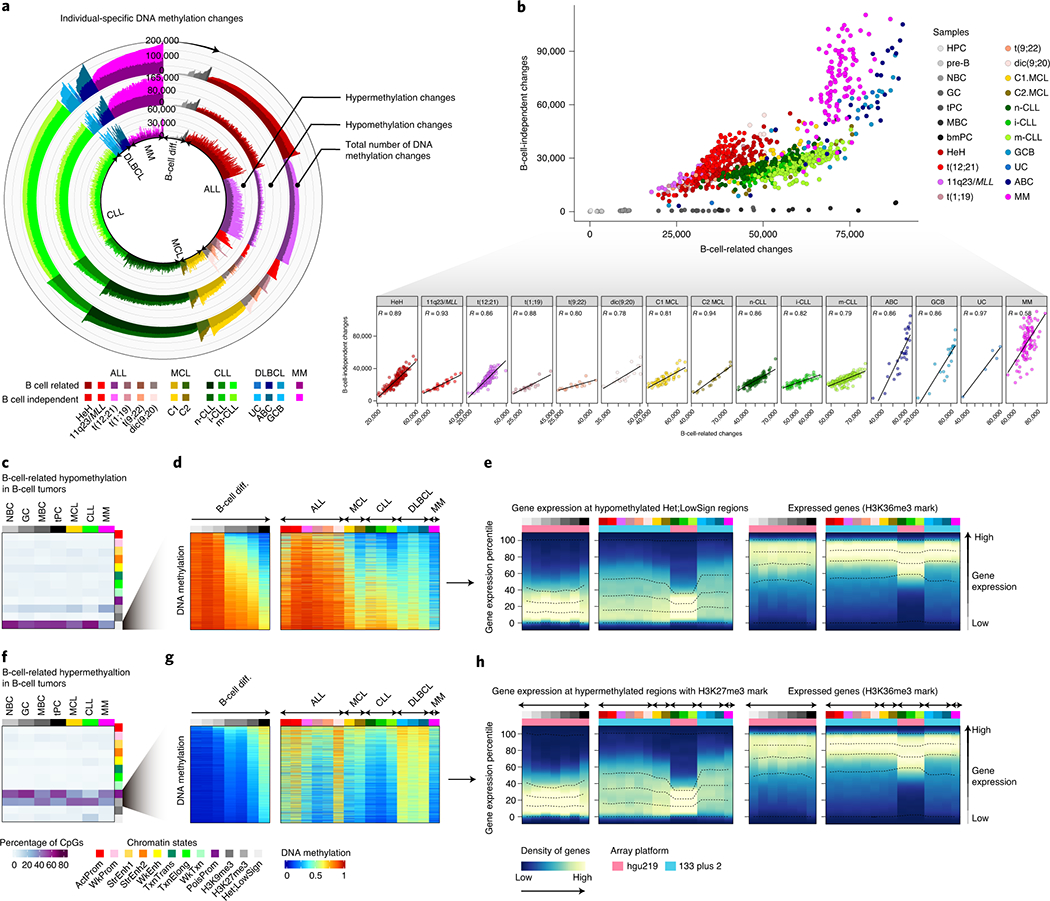
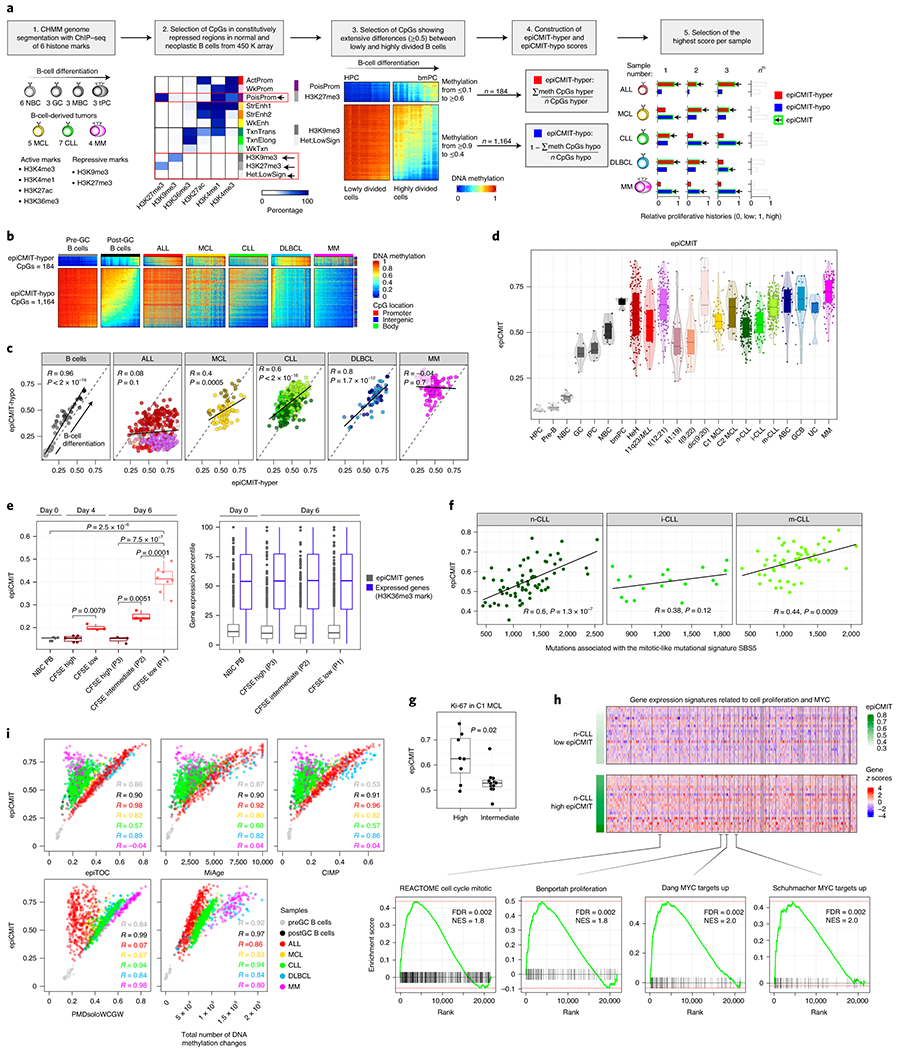
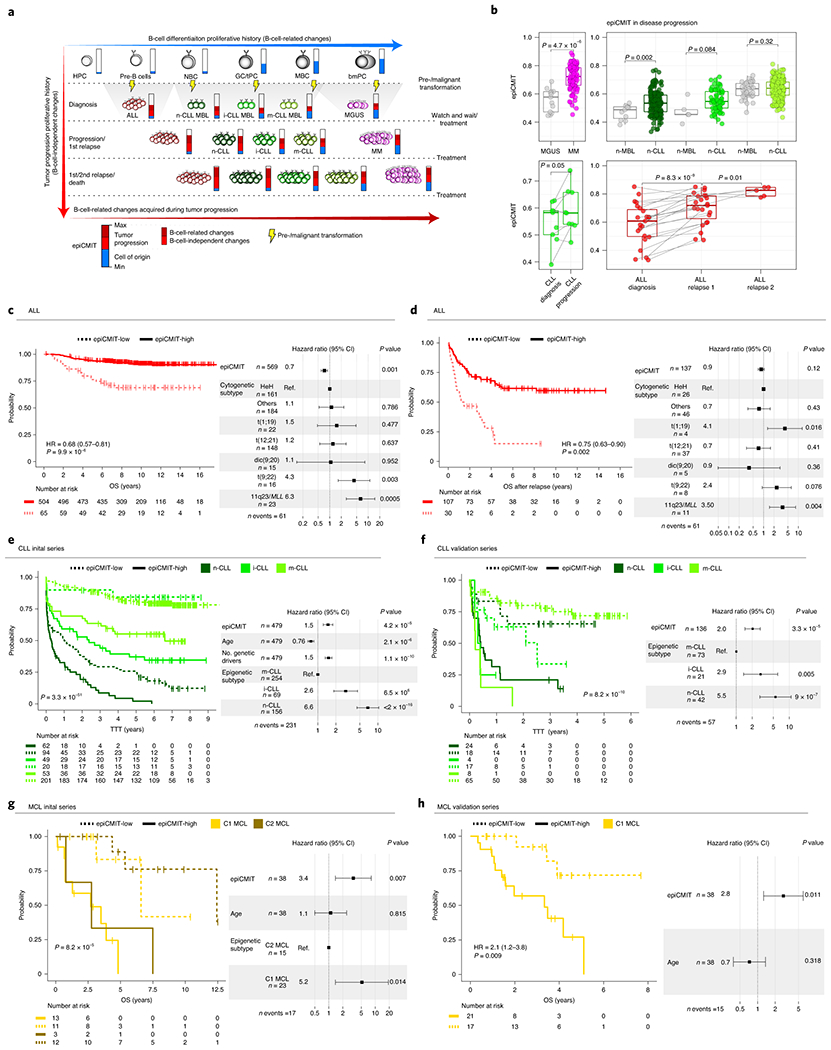
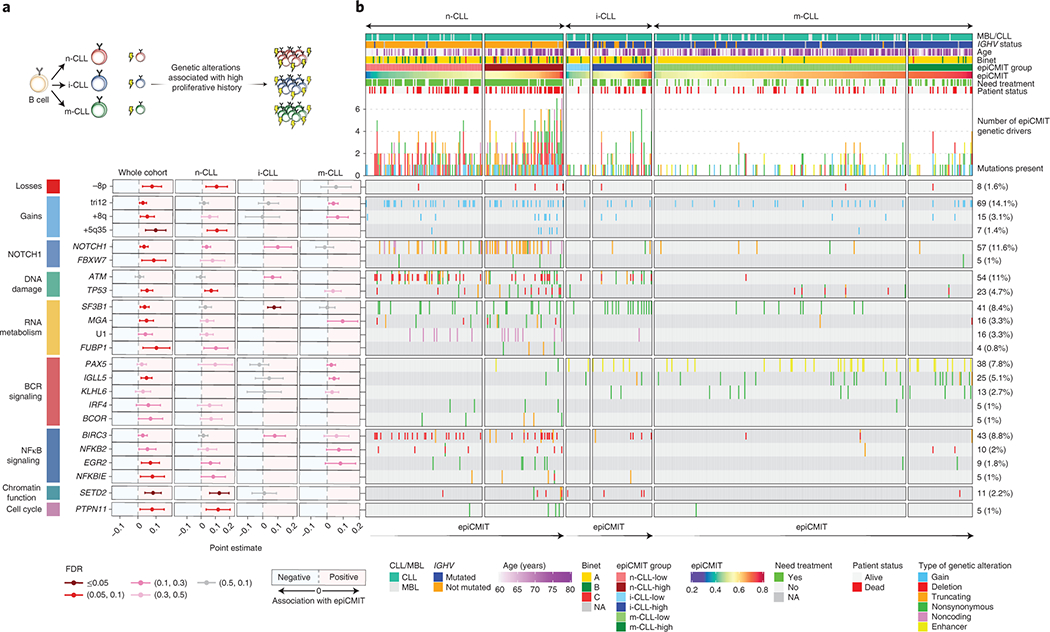
We report a systematic analysis of the DNA methylation variability in 1,595 samples of normal cell subpopulations and 14 tumor subtypes spanning the entire human B-cell lineage. Differential methylation among tumor entities relates to differences in cellular origin and to de novo epigenetic alterations, which allowed us to build an accurate machine learning-based diagnostic algorithm. We identify extensive patient-specific methylation variability in silenced chromatin associated with the proliferative history of normal and neoplastic B cells. Mitotic activity generally leaves both hyper- and hypomethylation imprints, but some B-cell neoplasms preferentially gain or lose DNA methylation. Subsequently, we construct a DNA methylation-based mitotic clock called epiCMIT, whose lapse magnitude represents a strong independent prognostic variable in B-cell tumors and is associated with particular driver genetic alterations. Our findings reveal DNA methylation as a holistic tracer of B-cell tumor developmental history, with implications in the differential diagnosis and prediction of clinical outcome.
Conflict of interest statement
COMPETING INTERESTS The authors declare no competing interests.
Figures

Comment in
-
A ticking clock for B cell tumors.Nat Cancer. 2020 Nov;1(11):1035-1037. doi: 10.1038/s43018-020-00132-1. Nat Cancer. 2020. PMID: 35122068 No abstract available.
Similar articles
-
An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells.Breast Cancer Res. 2016 Feb 29;18(1):27. doi: 10.1186/s13058-016-0685-5. Breast Cancer Res. 2016. PMID: 26923702 Free PMC article.
-
Biological, prognostic, and therapeutic impact of the epigenome in CLL.Semin Hematol. 2024 Jun;61(3):172-180. doi: 10.1053/j.seminhematol.2023.11.005. Epub 2023 Dec 1. Semin Hematol. 2024. PMID: 38151379 Review.
-
Epigenomic Characterization of Lymphoid Neoplasms.Annu Rev Pathol. 2024 Jan 24;19:371-396. doi: 10.1146/annurev-pathmechdis-051122-100856. Epub 2023 Oct 13. Annu Rev Pathol. 2024. PMID: 37832942 Review.
-
DNA methylation and cancer.Adv Genet. 2010;70:27-56. doi: 10.1016/B978-0-12-380866-0.60002-2. Adv Genet. 2010. PMID: 20920744 Review.
-
An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance.Theranostics. 2021 Mar 11;11(11):5346-5364. doi: 10.7150/thno.58385. eCollection 2021. Theranostics. 2021. PMID: 33859751 Free PMC article.
Cited by
-
Celebrating Ulrik Ringborg: Multi-Omics-Based Patient Stratification for Precision Cancer Treatment.Biomolecules. 2025 May 10;15(5):693. doi: 10.3390/biom15050693. Biomolecules. 2025. PMID: 40427586 Free PMC article. Review.
-
Aging and cancer epigenetics: Where do the paths fork?Aging Cell. 2022 Oct;21(10):e13709. doi: 10.1111/acel.13709. Epub 2022 Sep 14. Aging Cell. 2022. PMID: 36103298 Free PMC article. Review.
-
Converging genetic and epigenetic drivers of paediatric acute lymphoblastic leukaemia identified by an information-theoretic analysis.Nat Biomed Eng. 2021 Apr;5(4):360-376. doi: 10.1038/s41551-021-00703-2. Epub 2021 Apr 15. Nat Biomed Eng. 2021. PMID: 33859388 Free PMC article.
-
5-methylcytosine turnover: Mechanisms and therapeutic implications in cancer.Front Mol Biosci. 2022 Aug 17;9:976862. doi: 10.3389/fmolb.2022.976862. eCollection 2022. Front Mol Biosci. 2022. PMID: 36060265 Free PMC article. Review.
-
More than bad luck: Cancer and aging are linked to replication-driven changes to the epigenome.Sci Adv. 2023 Jul 21;9(29):eadf4163. doi: 10.1126/sciadv.adf4163. Epub 2023 Jul 19. Sci Adv. 2023. PMID: 37467337 Free PMC article.
References
-
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, J. T WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (International Agency for Research on Cancer (IARC), 2017).
METHODS REFERENCES
-
- Brönneke S et al. DNA methylation regulates lineage-specifying genes in primary lymphatic and blood endothelial cells. Angiogenesis 15, 317–329 (2012). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
