

Acute systemic inflammation exacerbates neuroinflammation in Alzheimer's disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction
- PMID: 34080771
- PMCID: PMC8874214
- DOI: 10.1002/alz.12341
Acute systemic inflammation exacerbates neuroinflammation in Alzheimer's disease: IL-1β drives amplified responses in primed astrocytes and neuronal network dysfunction
Abstract
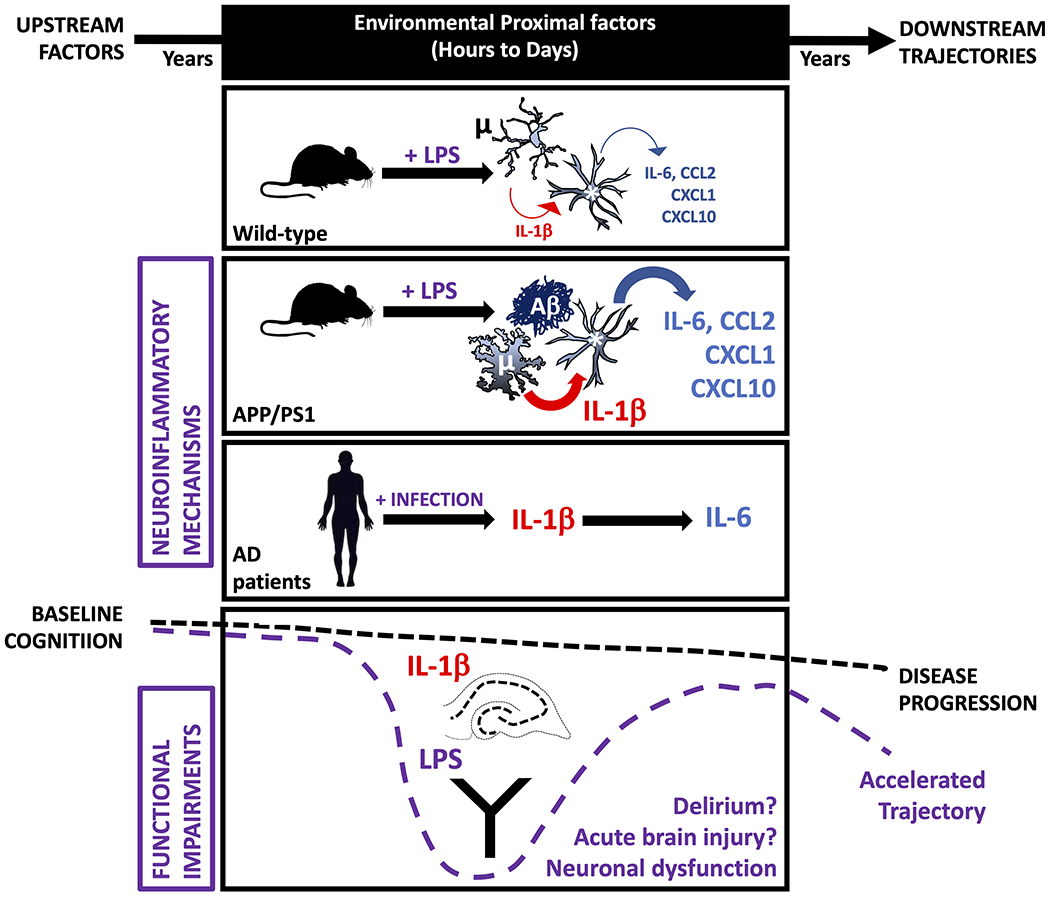
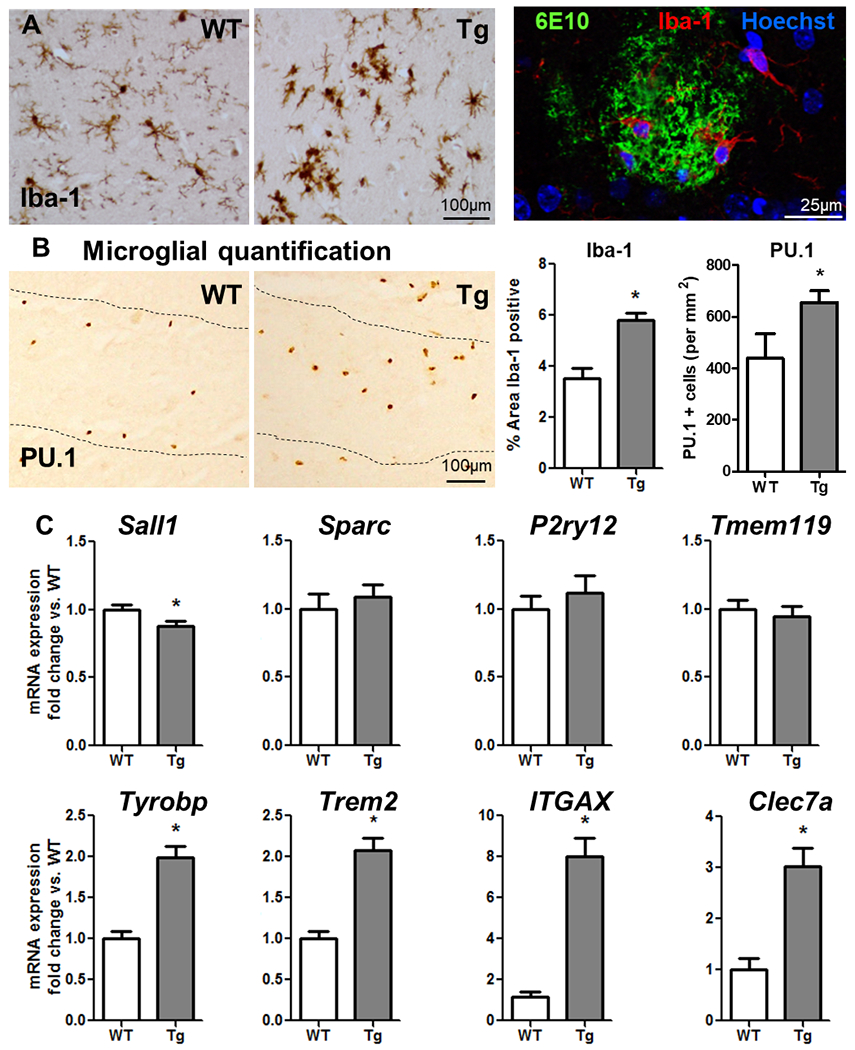
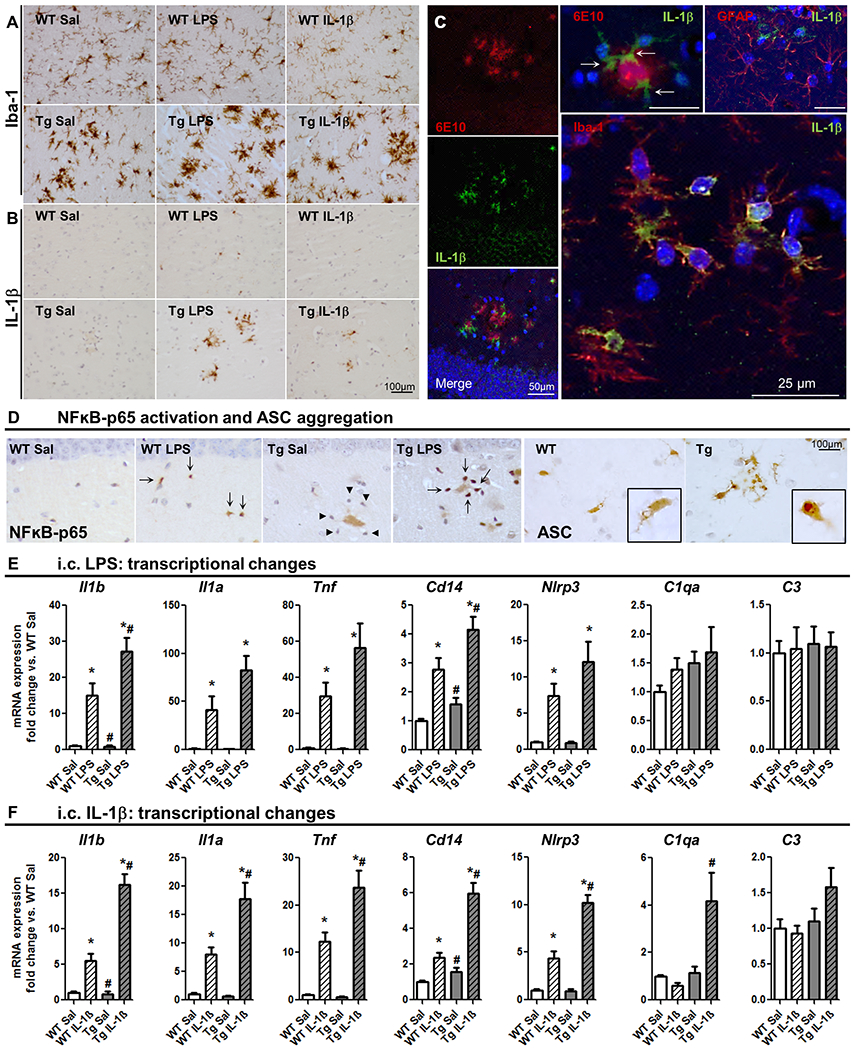
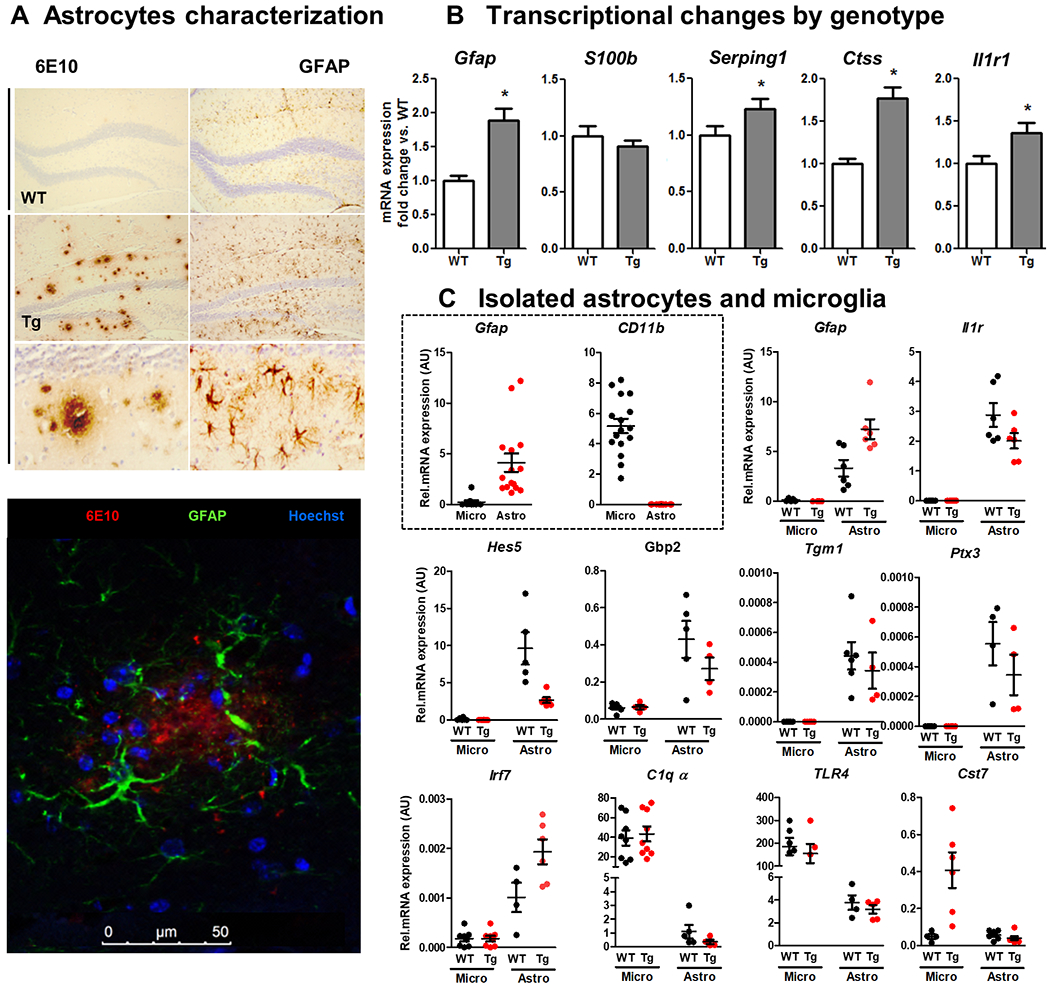
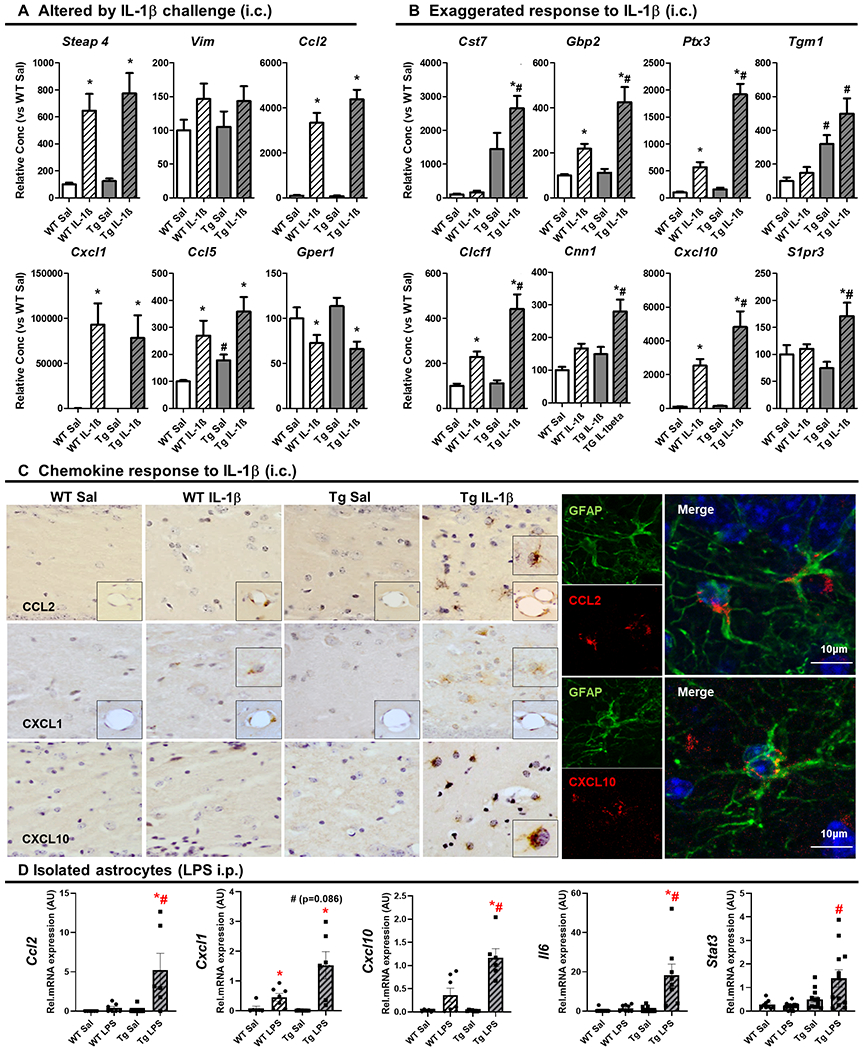
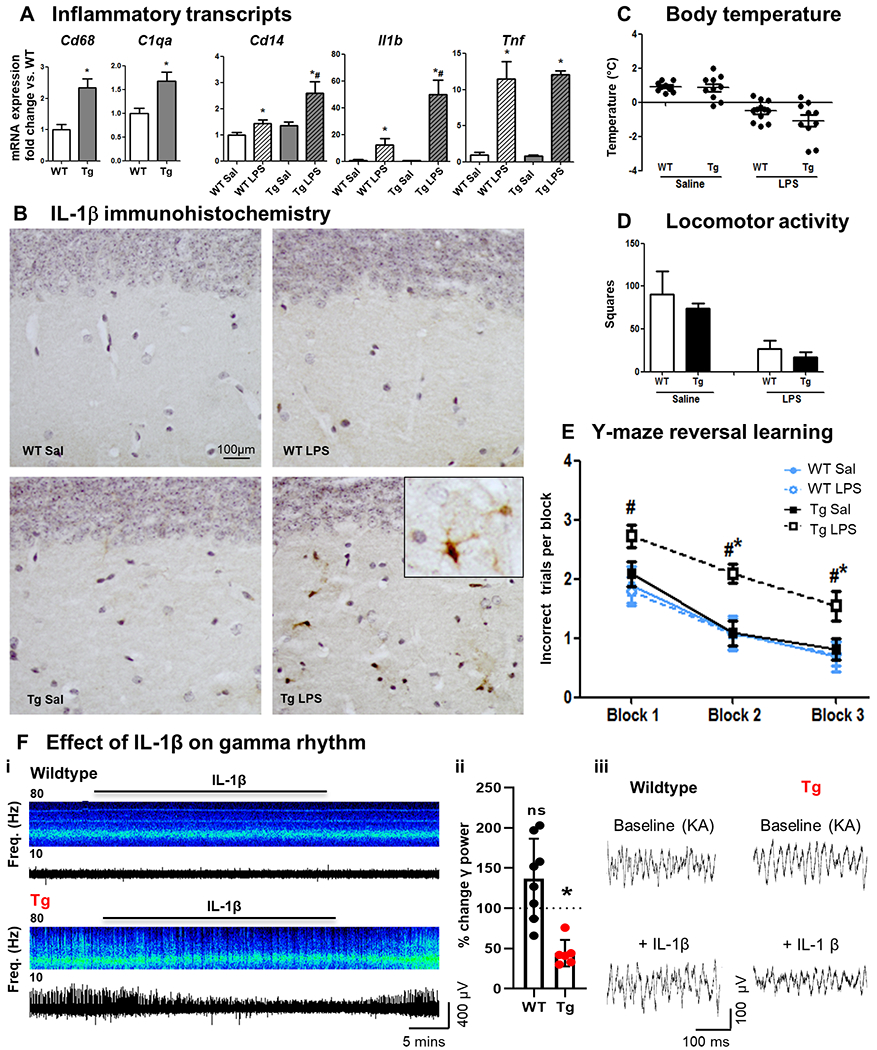
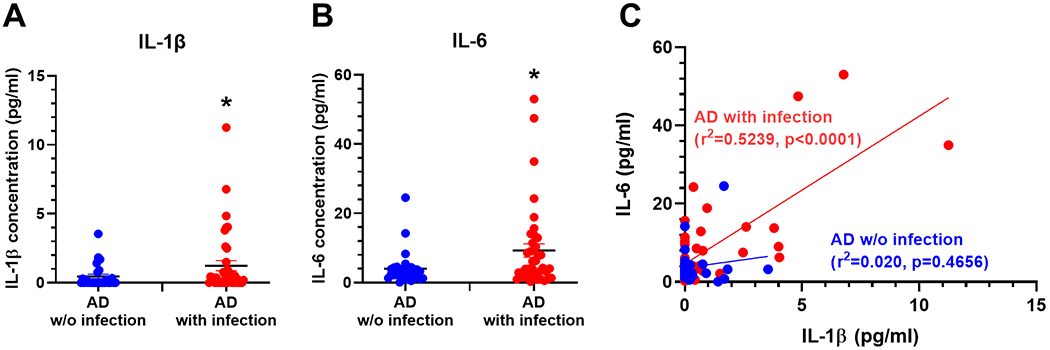
Neuroinflammation contributes to Alzheimer's disease (AD) progression. Secondary inflammatory insults trigger delirium and can accelerate cognitive decline. Individual cellular contributors to this vulnerability require elucidation. Using APP/PS1 mice and AD brain, we studied secondary inflammatory insults to investigate hypersensitive responses in microglia, astrocytes, neurons, and human brain tissue. The NLRP3 inflammasome was assembled surrounding amyloid beta, and microglia were primed, facilitating exaggerated interleukin-1β (IL-1β) responses to subsequent LPS stimulation. Astrocytes were primed to produce exaggerated chemokine responses to intrahippocampal IL-1β. Systemic LPS triggered microglial IL-1β, astrocytic chemokines, IL-6, and acute cognitive dysfunction, whereas IL-1β disrupted hippocampal gamma rhythm, all selectively in APP/PS1 mice. Brains from AD patients with infection showed elevated IL-1β and IL-6 levels. Therefore, amyloid leaves the brain vulnerable to secondary inflammation at microglial, astrocytic, neuronal, and cognitive levels, and infection amplifies neuroinflammatory cytokine synthesis in humans. Exacerbation of neuroinflammation to produce deleterious outcomes like delirium and accelerated disease progression merits careful investigation in humans.
Keywords: APP/PS1; CCL2; IL-1β; astrocyte; chemokine; cytokine; delirium; dementia; gamma; memory; microglia; network dysfunction; neuroinflammation; primed; priming; vulnerability.
© 2021 The Authors. Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
Figures
References
-
- Clark CM, Sheppard L, Fillenbaum GG, et al. Variability in annual Mini-Mental State Examination score in patients with probable Alzheimer disease: a clinical perspective of data from the Consortium to Establish a Registry for Alzheimer’s Disease. Arch Neurol. 1999;56(7):857–862. - PubMed
-
- Holmes C, Lovestone S. Long-term cognitive and functional decline in late onset Alzheimer’s disease: therapeutic implications. Age Ageing. 2003;32(2):200–204. - PubMed
-
- Semmler A, Widmann CN, Okulla T, et al. Persistent cognitive impairment, hippocampal atrophy and EEG changes in sepsis survivors. J Neurol Neurosurg Psychiatry. 2013;84(1):62–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
