

Cotton D genome assemblies built with long-read data unveil mechanisms of centromere evolution and stress tolerance divergence
- PMID: 34082735
- PMCID: PMC8176745
- DOI: 10.1186/s12915-021-01041-0
Cotton D genome assemblies built with long-read data unveil mechanisms of centromere evolution and stress tolerance divergence
Abstract
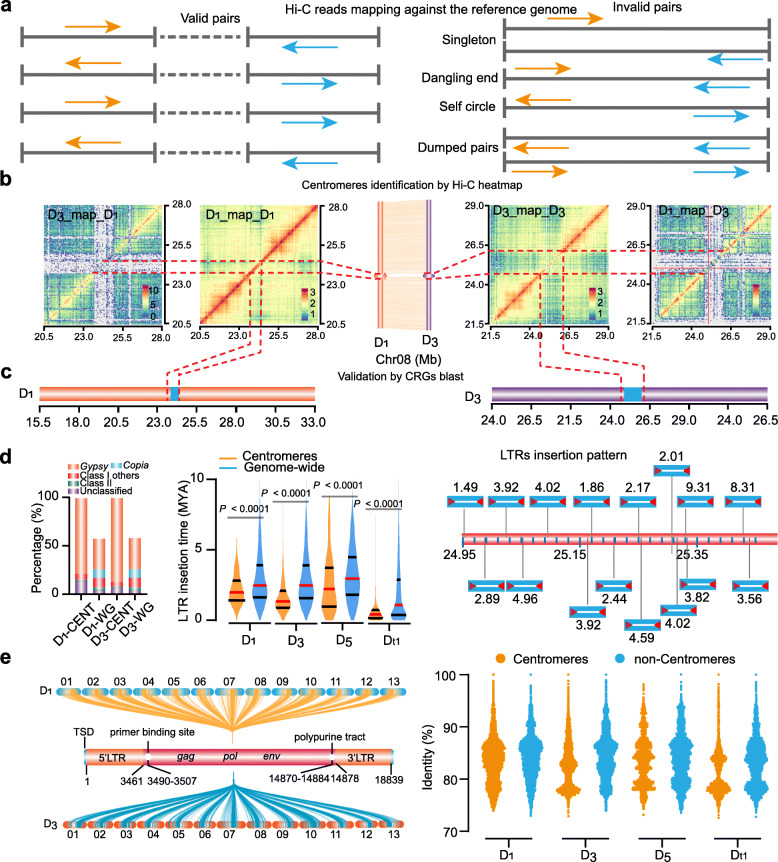
Background: Many of genome features which could help unravel the often complex post-speciation evolution of closely related species are obscured because of their location in chromosomal regions difficult to accurately characterize using standard genome analysis methods, including centromeres and repeat regions.
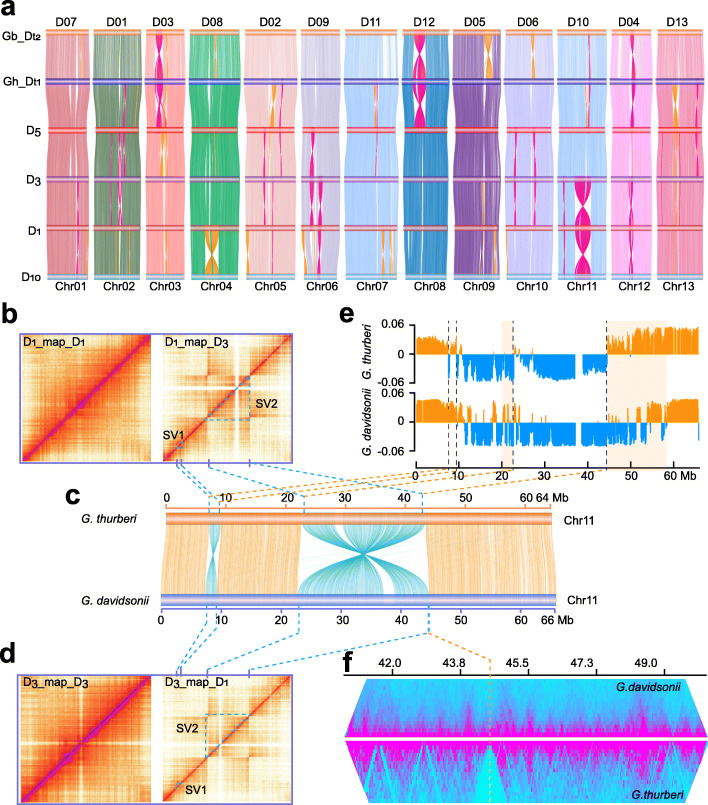
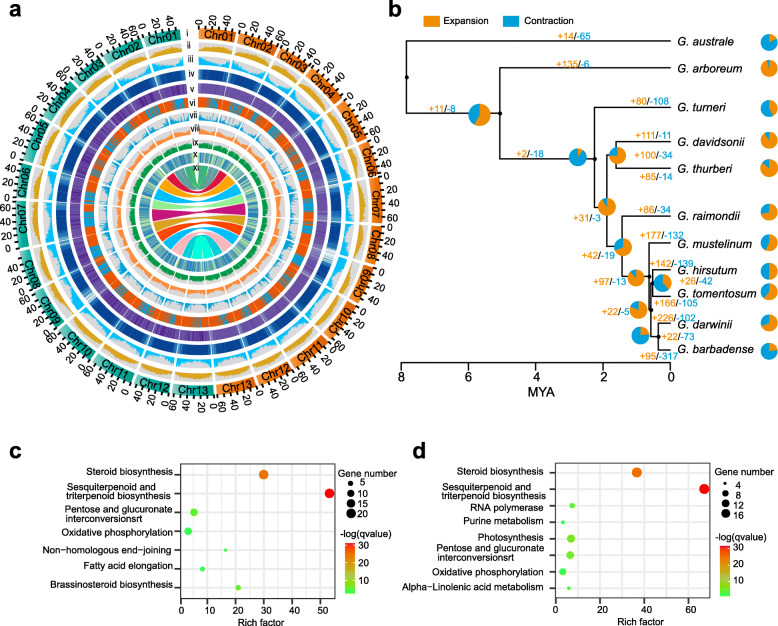
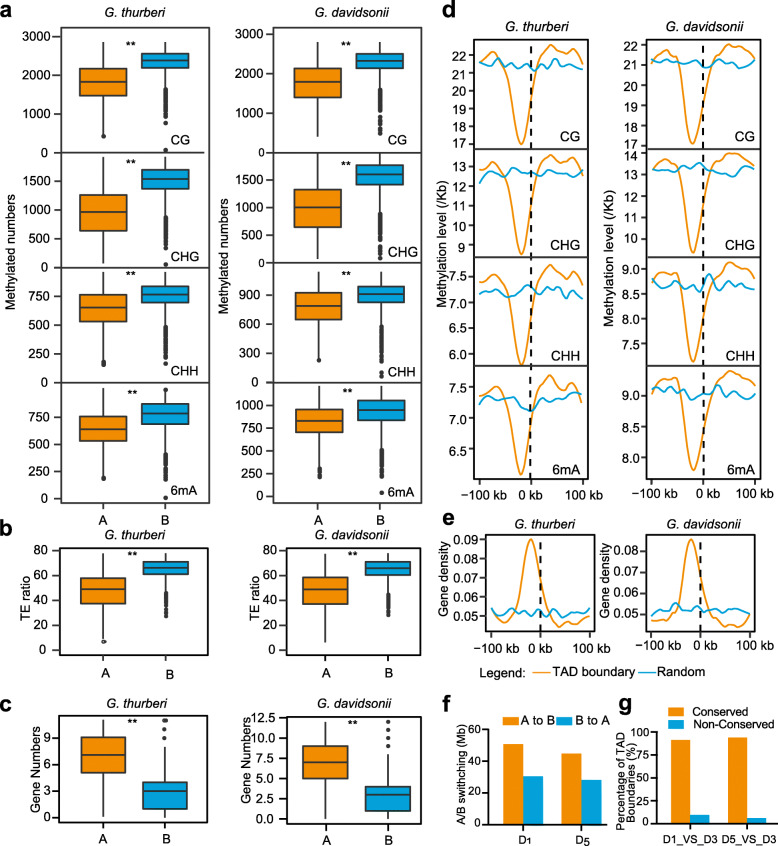
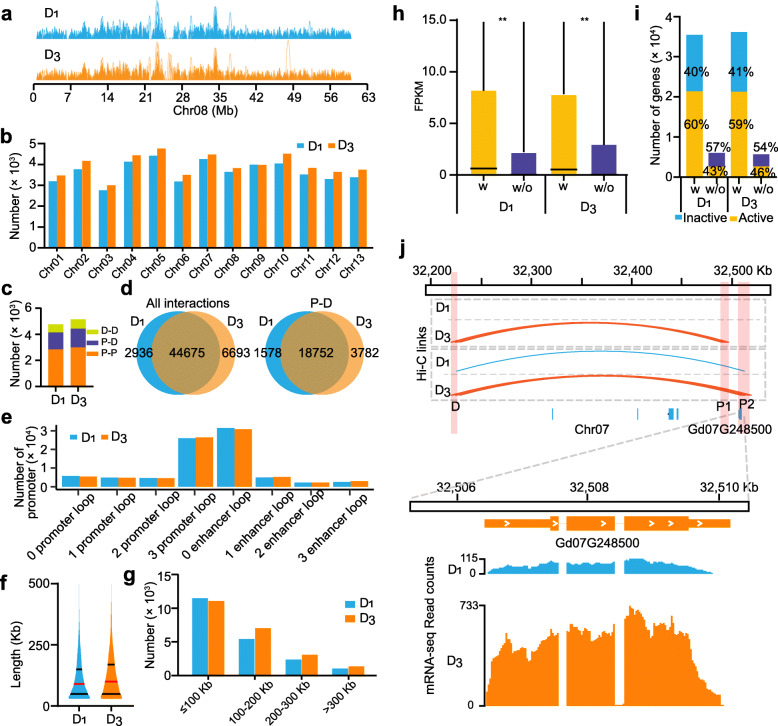
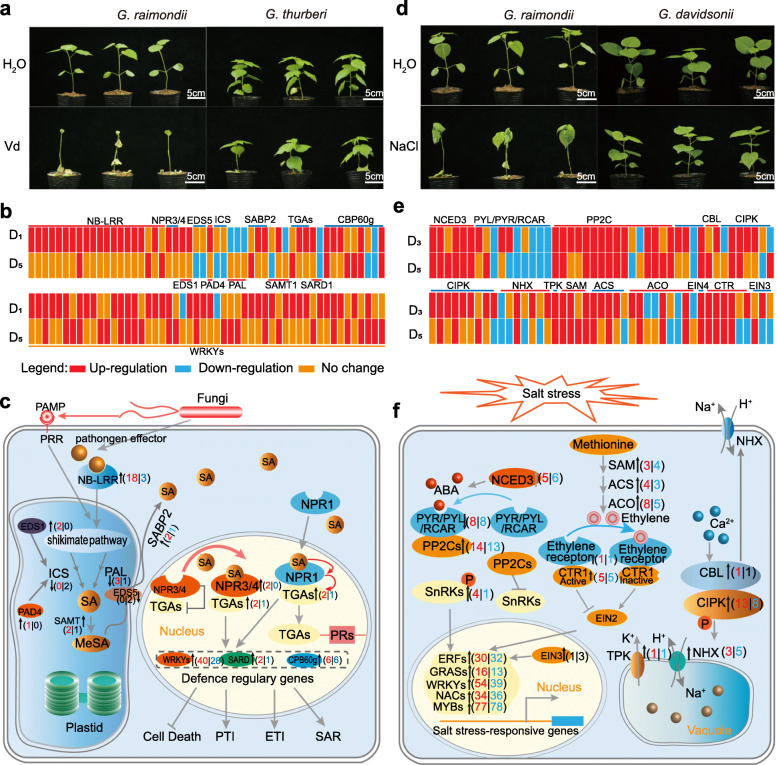
Results: Here, we analyze the genome evolution and diversification of two recently diverged sister cotton species based on nanopore long-read sequence assemblies and Hi-C 3D genome data. Although D genomes are conserved in gene content, they have diversified in gene order, gene structure, gene family diversification, 3D chromatin structure, long-range regulation, and stress-related traits. Inversions predominate among D genome rearrangements. Our results support roles for 5mC and 6mA in gene activation, and 3D chromatin analysis showed that diversification in proximal-vs-distal regulatory-region interactions shape the regulation of defense-related-gene expression. Using a newly developed method, we accurately positioned cotton centromeres and found that these regions have undergone obviously more rapid evolution relative to chromosome arms. We also discovered a cotton-specific LTR class that clarifies evolutionary trajectories among diverse cotton species and identified genetic networks underlying the Verticillium tolerance of Gossypium thurberi (e.g., SA signaling) and salt-stress tolerance of Gossypium davidsonii (e.g., ethylene biosynthesis). Finally, overexpression of G. thurberi genes in upland cotton demonstrated how wild cottons can be exploited for crop improvement.
Conclusions: Our study substantially deepens understanding about how centromeres have developed and evolutionarily impacted the divergence among closely related cotton species and reveals genes and 3D genome structures which can guide basic investigations and applied efforts to improve crops.
Keywords: 3D genome; G. davidsonii; G. thurberi; Gossypium; Hi-C; Long-range interactions; Salt tolerance; Structural variation; Verticillium wilt.
Conflict of interest statement
The authors declare that there are no competing interests.
Figures
Similar articles
-
High-quality Gossypium hirsutum and Gossypium barbadense genome assemblies reveal the landscape and evolution of centromeres.Plant Commun. 2024 Feb 12;5(2):100722. doi: 10.1016/j.xplc.2023.100722. Epub 2023 Sep 22. Plant Commun. 2024. PMID: 37742072 Free PMC article.
-
Comparative transcriptome analysis reveals evolutionary divergence and shared network of cold and salt stress response in diploid D-genome cotton.BMC Plant Biol. 2020 Nov 12;20(1):518. doi: 10.1186/s12870-020-02726-4. BMC Plant Biol. 2020. PMID: 33183239 Free PMC article.
-
Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement.Nat Genet. 2020 May;52(5):525-533. doi: 10.1038/s41588-020-0614-5. Epub 2020 Apr 20. Nat Genet. 2020. PMID: 32313247 Free PMC article.
-
Transposable elements play an important role during cotton genome evolution and fiber cell development.Sci China Life Sci. 2016 Feb;59(2):112-21. doi: 10.1007/s11427-015-4928-y. Epub 2015 Dec 19. Sci China Life Sci. 2016. PMID: 26687725 Review.
-
Deciphering the complex cotton genome for improving fiber traits and abiotic stress resilience in sustainable agriculture.Mol Biol Rep. 2023 Aug;50(8):6937-6953. doi: 10.1007/s11033-023-08565-4. Epub 2023 Jun 22. Mol Biol Rep. 2023. PMID: 37349608 Review.
Cited by
-
Assembly and annotation of the Gossypium barbadense L. 'Pima-S6' genome raise questions about the chromosome structure and gene content of Gossypium barbadense genomes.BMC Genomics. 2023 Jan 10;24(1):11. doi: 10.1186/s12864-022-09102-6. BMC Genomics. 2023. PMID: 36627552 Free PMC article.
-
Non-B-form DNA tends to form in centromeric regions and has undergone changes in polyploid oat subgenomes.Proc Natl Acad Sci U S A. 2023 Jan 3;120(1):e2211683120. doi: 10.1073/pnas.2211683120. Epub 2022 Dec 27. Proc Natl Acad Sci U S A. 2023. PMID: 36574697 Free PMC article.
-
Regulatory networks of coresident subgenomes during rapid fiber cell elongation in upland cotton.Plant Commun. 2024 Dec 9;5(12):101130. doi: 10.1016/j.xplc.2024.101130. Epub 2024 Sep 10. Plant Commun. 2024. PMID: 39257006 Free PMC article.
-
Centromere-Specific Retrotransposons and Very-Long-Chain Fatty Acid Biosynthesis in the Genome of Yellowhorn (Xanthoceras sorbifolium, Sapindaceae), an Oil-Producing Tree With Significant Drought Resistance.Front Plant Sci. 2021 Nov 22;12:766389. doi: 10.3389/fpls.2021.766389. eCollection 2021. Front Plant Sci. 2021. PMID: 34880890 Free PMC article.
-
Two large inversions seriously suppress recombination and are essential for key genotype fixation in cabbage (Brassica oleracea L. var. capitata).Hortic Res. 2024 Jan 30;11(4):uhae030. doi: 10.1093/hr/uhae030. eCollection 2024 Apr. Hortic Res. 2024. PMID: 39896709 Free PMC article.
References
-
- Mehboob ur R, Shaheen T, Tabbasam N, Iqbal MA, Ashraf M, Zafar Y, Paterson AH. Cotton genetic resources. A review. Agronomy Sustainable Dev. 2011;32:419–432. doi: 10.1007/s13593-011-0051-z. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
