

Birth prevalence of phenylalanine hydroxylase deficiency: a systematic literature review and meta-analysis
- PMID: 34082800
- PMCID: PMC8173927
- DOI: 10.1186/s13023-021-01874-6
Birth prevalence of phenylalanine hydroxylase deficiency: a systematic literature review and meta-analysis
Abstract
Background: Phenylalanine hydroxylase (PAH) deficiency is an autosomal recessive disorder that results in elevated concentrations of phenylalanine (Phe) in the blood. If left untreated, the accumulation of Phe can result in profound neurocognitive disability. The objective of this systematic literature review and meta-analysis was to estimate the global birth prevalence of PAH deficiency from newborn screening studies and to estimate regional differences, overall and for various clinically relevant Phe cutoff values used in confirmatory testing.
Methods: The protocol for this literature review was registered with PROSPERO (International prospective register of systematic reviews). Pubmed and Embase database searches were used to identify studies that reported the birth prevalence of PAH deficiency. Only studies including numeric birth prevalence reports of confirmed PAH deficiency were included.
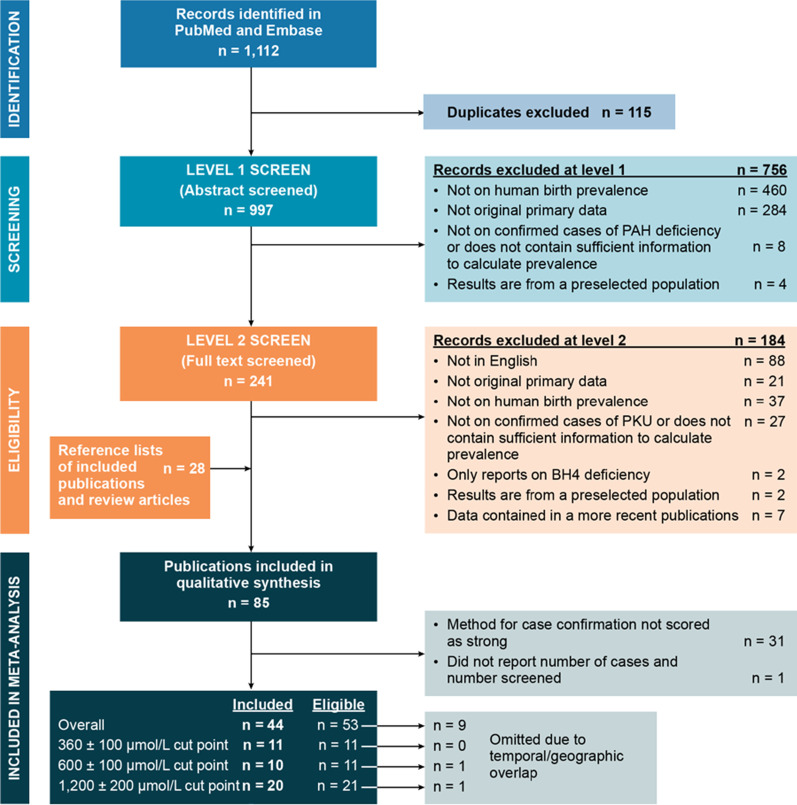
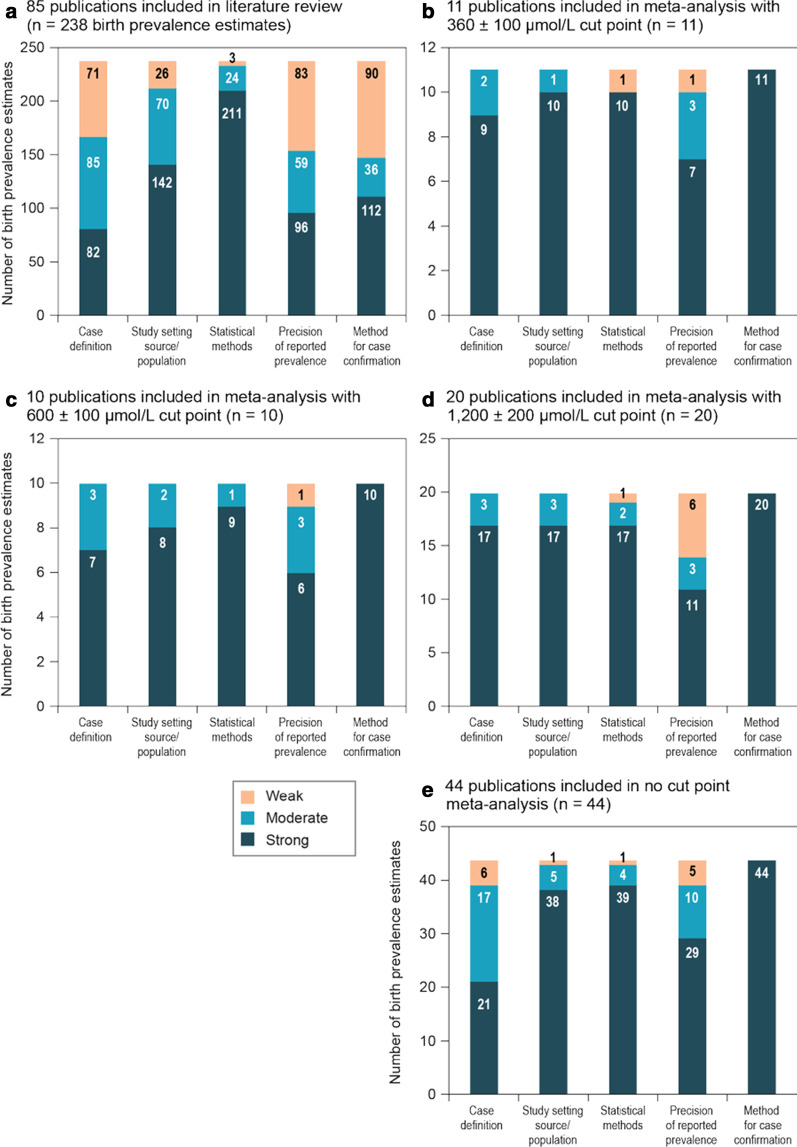
Results: From the 85 publications included in the review, 238 birth prevalence estimates were extracted. After excluding prevalence estimates that did not meet quality assessment criteria or because of temporal and regional overlap, estimates from 45 publications were included in the meta-analysis. The global birth prevalence of PAH deficiency, estimated by weighting regional birth prevalences relative to their share of the population of all regions included in the study, was 0.64 (95% confidence interval [CI] 0.53-0.75) per 10,000 births and ranged from 0.03 (95% CI 0.02-0.05) per 10,000 births in Southeast Asia to 1.18 (95% CI 0.64-1.87) per 10,000 births in the Middle East/North Africa. Regionally weighted global birth prevalences per 10,000 births by confirmatory test Phe cutoff values were 0.96 (95% CI 0.50-1.42) for the Phe cutoff value of 360 ± 100 µmol/L; 0.50 (95% CI 0.37-0.64) for the Phe cutoff value of 600 ± 100 µmol/L; and 0.30 (95% CI 0.20-0.40) for the Phe cutoff value of 1200 ± 200 µmol/L.
Conclusions: Substantial regional variation in the birth prevalence of PAH deficiency was observed in this systematic literature review and meta-analysis of published evidence from newborn screening. The precision of the prevalence estimates is limited by relatively small sample sizes, despite widespread and longstanding newborn screening in much of the world.
Keywords: Hyperphenylalaninemia; Newborn screening; Phenylalanine hydroxylase deficiency; Phenylketonuria; Prevalence.
Conflict of interest statement
RS, KA, and SL are employees of BioMarin Pharmaceutical Inc. PKF was a consultant for BioMarin Pharmaceutical Inc. when this research was conducted. Research team members AH, AVM, BC and MP are full-time employees of RTI Health Solutions. RTI Health Solutions is a unit of RTI International, an independent, nonprofit organization that conducts work for government, public, and private organizations, including pharmaceutical companies. RTI authors participate in this work in the course of employment as work for hire, pursuant to a contract to conduct an independent research study for a client (BioMarin Pharmaceutical Inc.).
Figures
References
-
- PAHvdb: Phenylalanine Hydroxylase Gene Locus-Specific Database. 2020. http://www.biopku.org/home/pah.asp. Accessed 4 Sept 2020.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
