

Crowdsourced mapping of unexplored target space of kinase inhibitors
- PMID: 34083538
- PMCID: PMC8175708
- DOI: 10.1038/s41467-021-23165-1
Crowdsourced mapping of unexplored target space of kinase inhibitors
Abstract
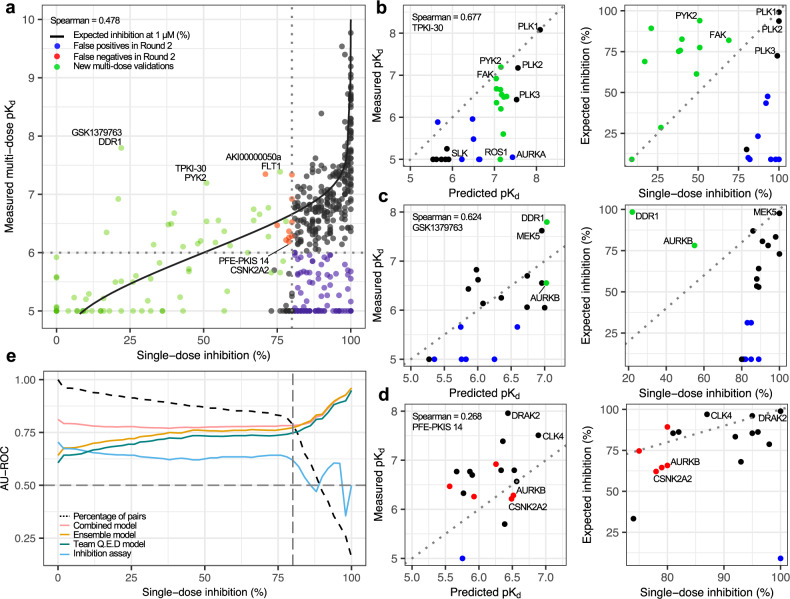
Despite decades of intensive search for compounds that modulate the activity of particular protein targets, a large proportion of the human kinome remains as yet undrugged. Effective approaches are therefore required to map the massive space of unexplored compound-kinase interactions for novel and potent activities. Here, we carry out a crowdsourced benchmarking of predictive algorithms for kinase inhibitor potencies across multiple kinase families tested on unpublished bioactivity data. We find the top-performing predictions are based on various models, including kernel learning, gradient boosting and deep learning, and their ensemble leads to a predictive accuracy exceeding that of single-dose kinase activity assays. We design experiments based on the model predictions and identify unexpected activities even for under-studied kinases, thereby accelerating experimental mapping efforts. The open-source prediction algorithms together with the bioactivities between 95 compounds and 295 kinases provide a resource for benchmarking prediction algorithms and for extending the druggable kinome.
Conflict of interest statement
The SGC is a registered charity that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (ULTRA-DD 115766), Wellcome Trust, Janssen, Merck Kga, Merck Sharp & Dohme, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, and Takeda. T.I.O. has received honoraria or consulted for Abbott, AstraZeneca, Chiron, Genentech, Infinity Pharmaceuticals, Merz Pharmaceuticals, Merck Darmstadt, Mitsubishi Tanabe, Novartis, Ono Pharmaceuticals, Pfizer, Roche, Sanofi and Wyeth. J.Z. is founder and CTO of Silexon AI Technology Co. Ltd. and has an equity interest. The rest of the authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
