

Interleukins in cancer: from biology to therapy
- PMID: 34083781
- PMCID: PMC8173513
- DOI: 10.1038/s41568-021-00363-z
Interleukins in cancer: from biology to therapy
Abstract
Interleukins and associated cytokines serve as the means of communication for innate and adaptive immune cells as well as non-immune cells and tissues. Thus, interleukins have a critical role in cancer development, progression and control. Interleukins can nurture an environment enabling and favouring cancer growth while simultaneously being essential for a productive tumour-directed immune response. These properties of interleukins can be exploited to improve immunotherapies to promote effectiveness as well as to limit side effects. This Review aims to unravel some of these complex interactions.
© 2021. Springer Nature Limited.
Conflict of interest statement
J.D. has received remuneration from Novartis for work unrelated to this Review. P.L. is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Novartis, Pfizer and Sanofi-Regeneron. P.L. is a member of scientific advisory board for Amgen, Caristo, Cartesian, Corvidia Therapeutics, CSL Behring, DalCor Pharmaceuticals, Dewpoint, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, PlaqueTec and XBiotech, Inc. P.L.’s laboratory has received research funding in the past 2 years from Novartis. P.L. is on the Board of Directors of XBiotech, and has a financial interest in Xbiotech, a company developing therapeutic human antibodies. P.L.’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict-of-interest policies. S.K. and S.E. are inventors named on several patents in the field of immuno-oncology unrelated to the present work. S.K. and S.E. received research support from TCR2 Inc. and Arcus Bioscience for work unrelated to this Review. S.K. and S.E. have licensed intellectual property to TCR2 Inc. S.K. has received honoraria from GlaxoSmithKline and Novartis. D.B. and C.A.D. declare no competing interests.
Figures
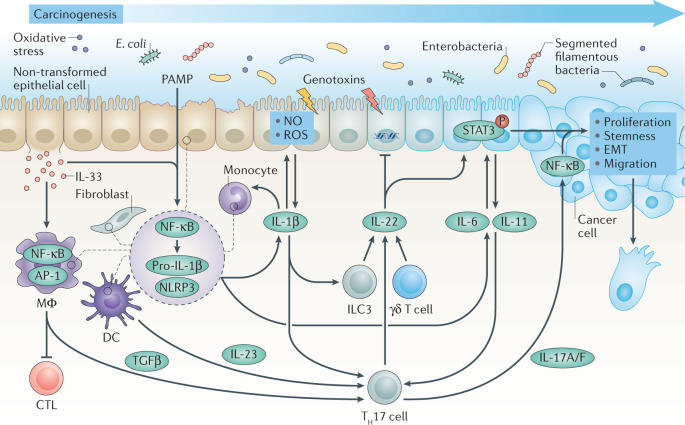
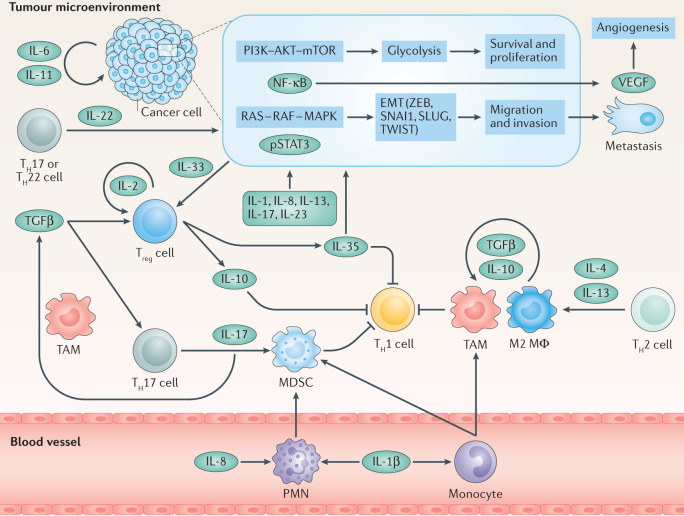
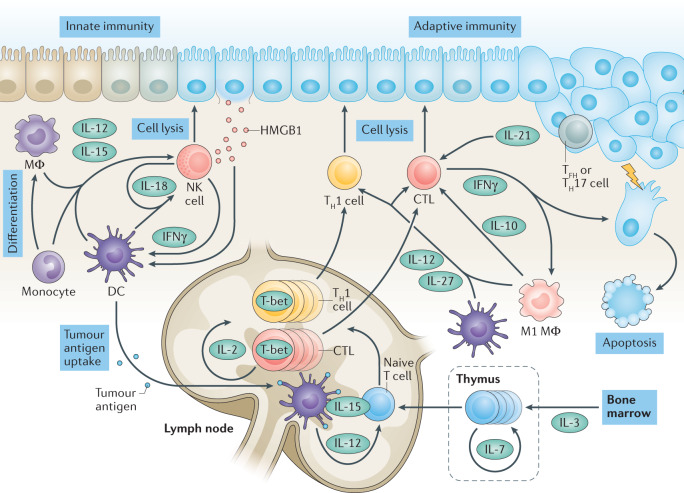
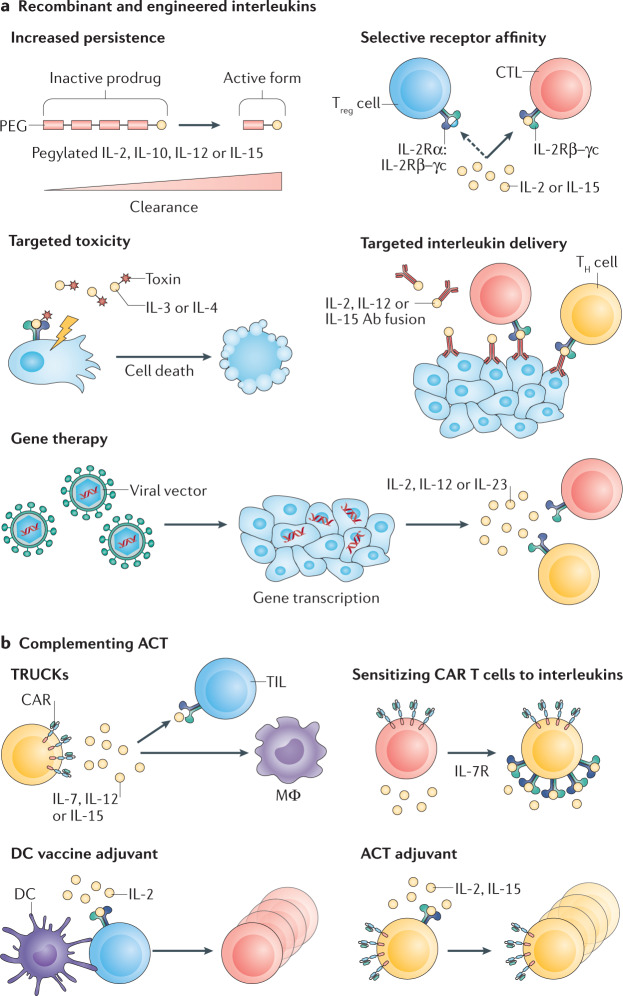
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
