

A map of transcriptional heterogeneity and regulatory variation in human microglia
- PMID: 34083789
- PMCID: PMC7610960
- DOI: 10.1038/s41588-021-00875-2
A map of transcriptional heterogeneity and regulatory variation in human microglia
Abstract
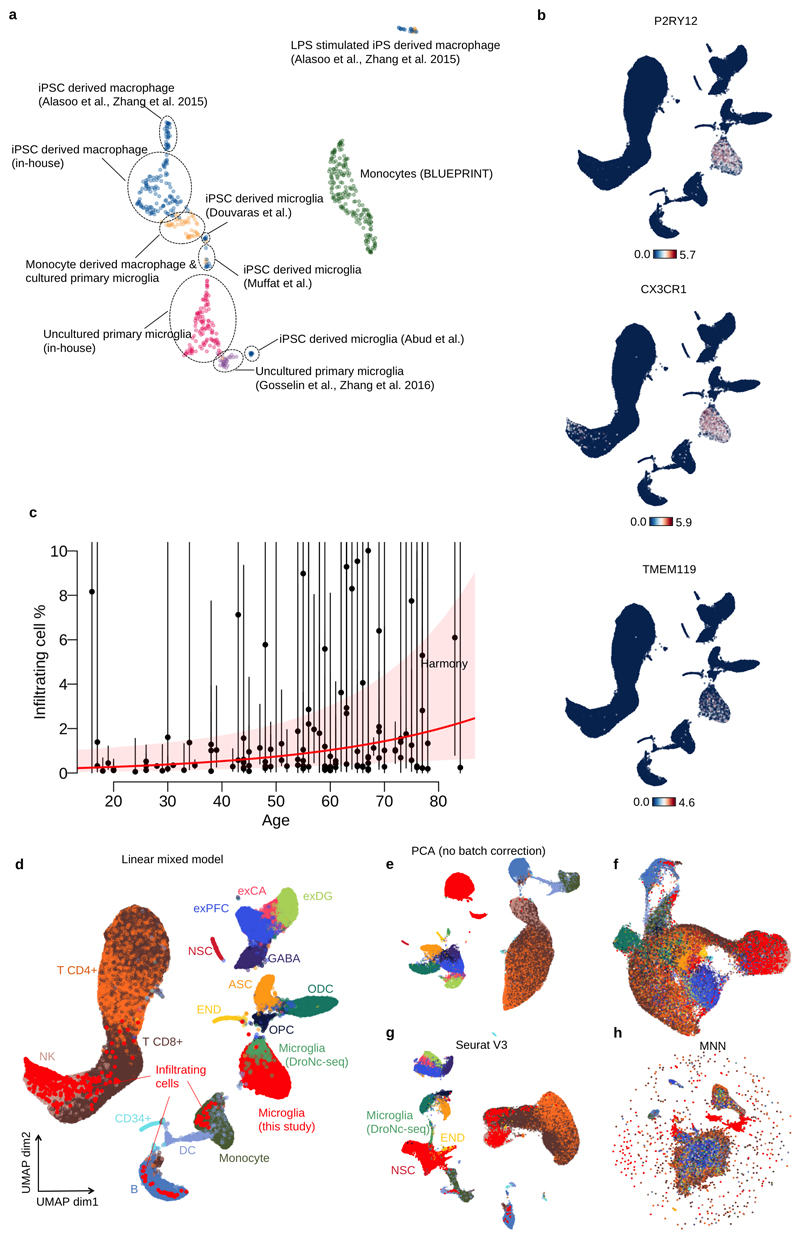
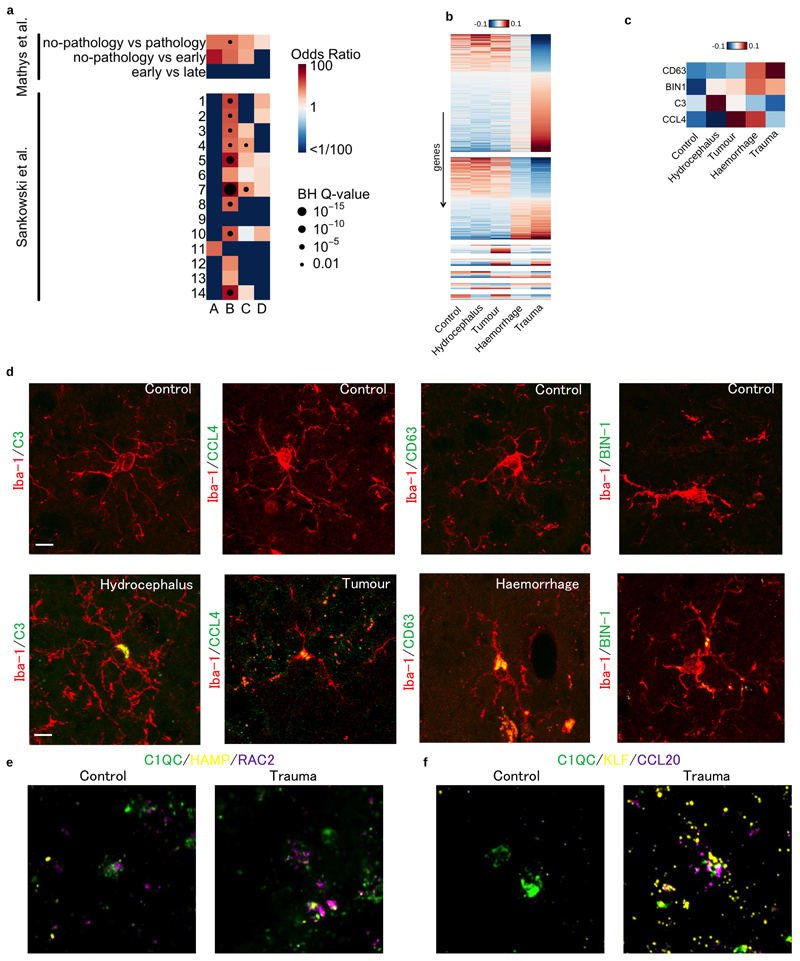
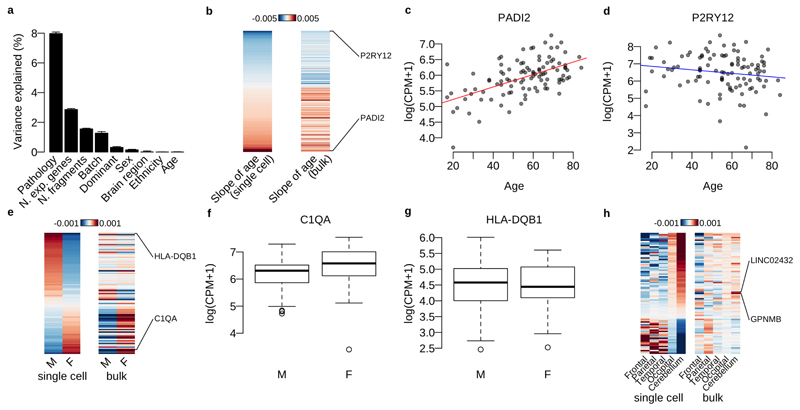
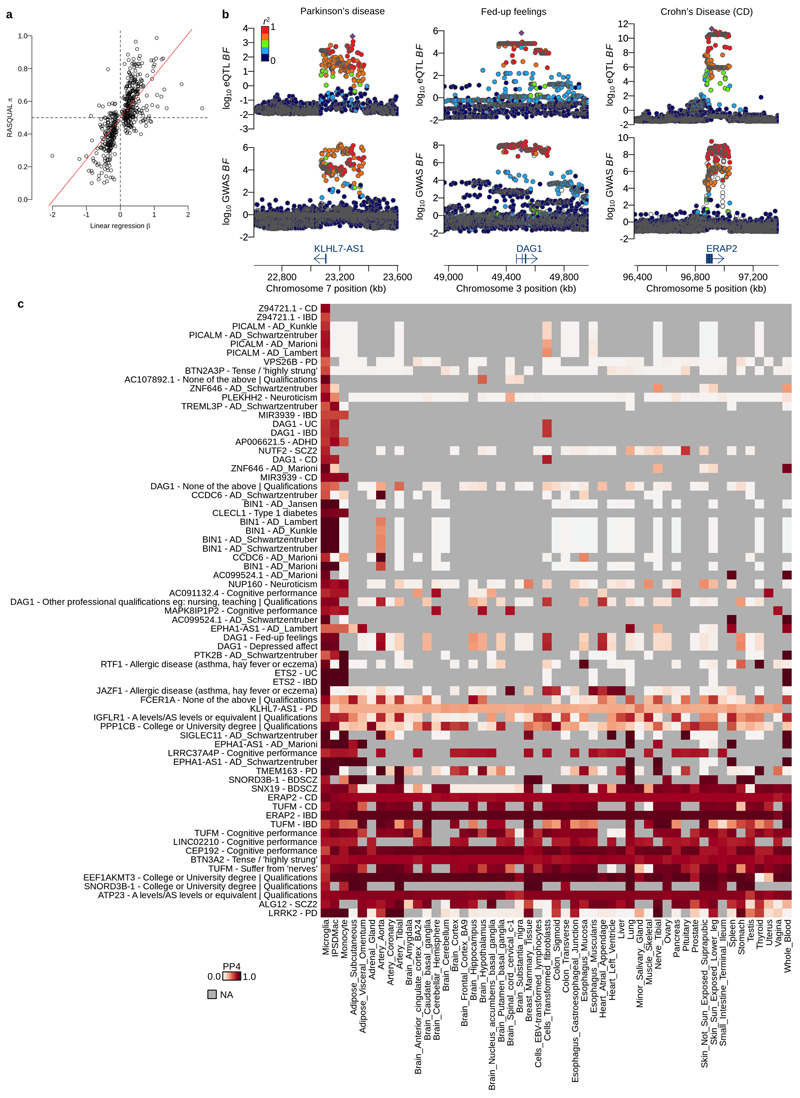
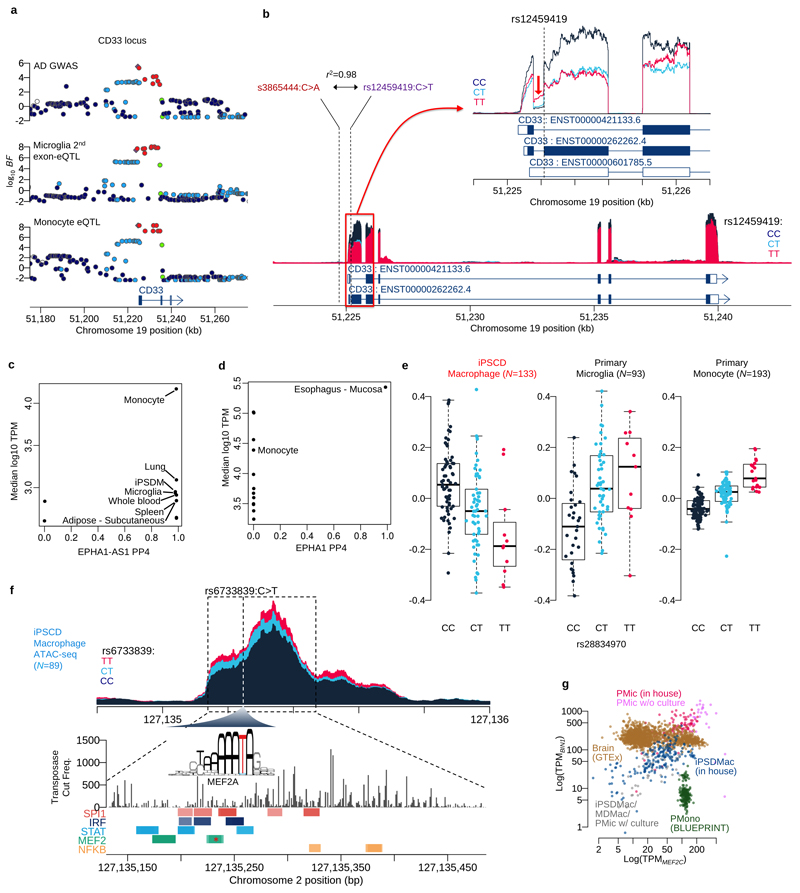
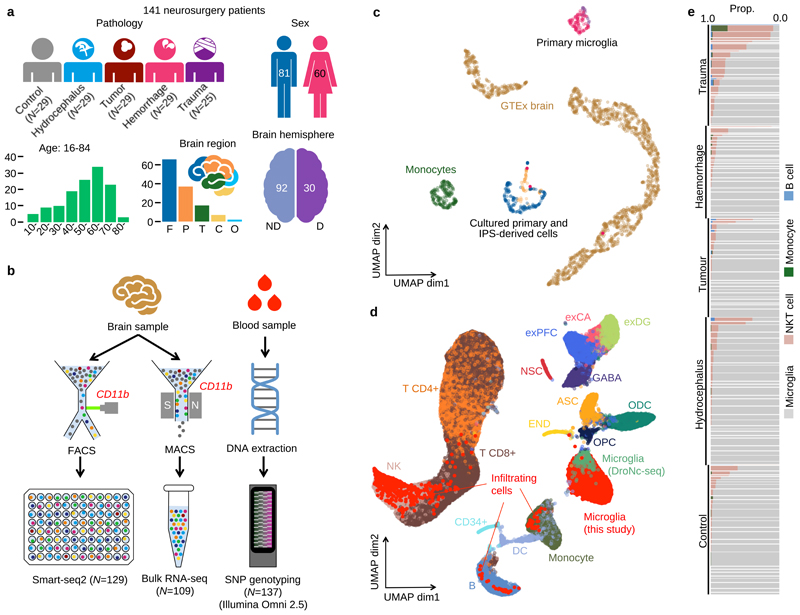
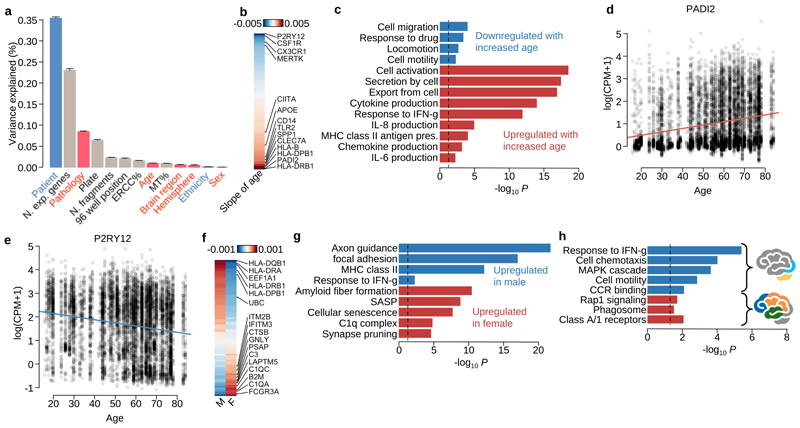
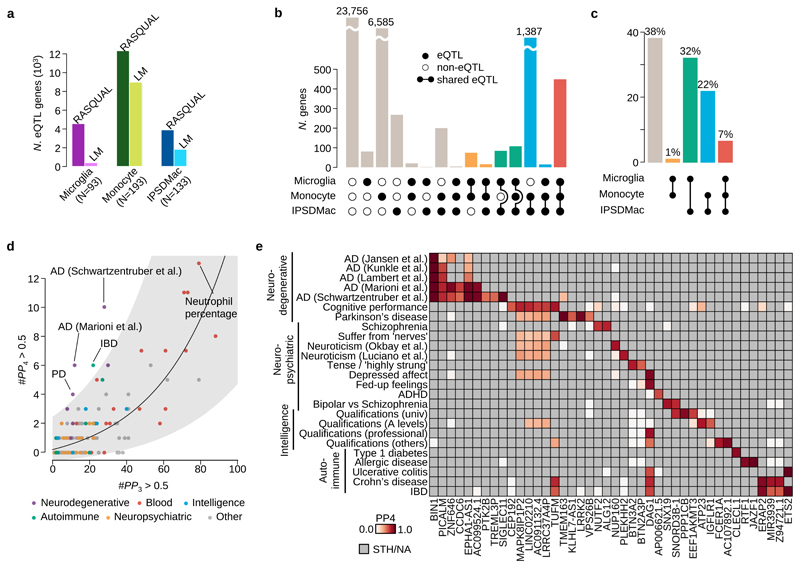
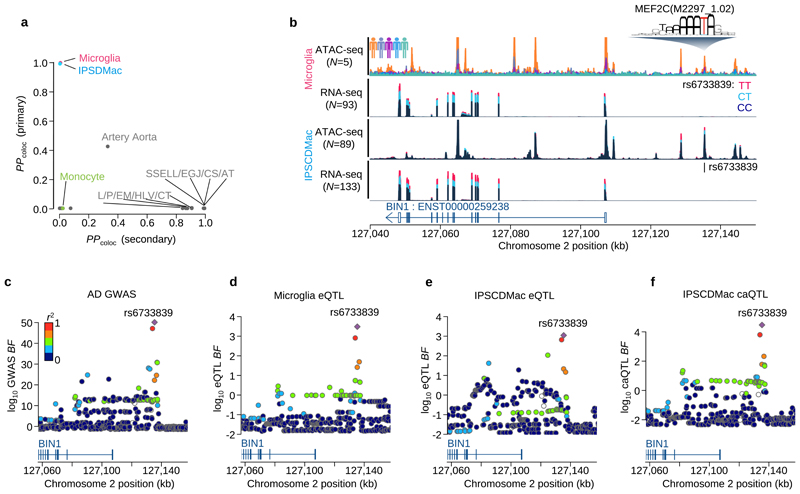
Microglia, the tissue-resident macrophages of the central nervous system (CNS), play critical roles in immune defense, development and homeostasis. However, isolating microglia from humans in large numbers is challenging. Here, we profiled gene expression variation in primary human microglia isolated from 141 patients undergoing neurosurgery. Using single-cell and bulk RNA sequencing, we identify how age, sex and clinical pathology influence microglia gene expression and which genetic variants have microglia-specific functions using expression quantitative trait loci (eQTL) mapping. We follow up one of our findings using a human induced pluripotent stem cell-based macrophage model to fine-map a candidate causal variant for Alzheimer's disease at the BIN1 locus. Our study provides a population-scale transcriptional map of a critically important cell for human CNS development and disease.
Conflict of interest statement
Competing Interests Statement
D.J.G. and E.M. were employees of Genomics PLC at the time the manuscript was submitted.
Figures

Comment in
-
Scrutinizing disease states and regulation in human microglia.Nat Genet. 2021 Jun;53(6):766-767. doi: 10.1038/s41588-021-00826-x. Nat Genet. 2021. PMID: 34083790 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
