

Phenotypic signatures in clinical data enable systematic identification of patients for genetic testing
- PMID: 34083811
- PMCID: PMC8981189
- DOI: 10.1038/s41591-021-01356-z
Phenotypic signatures in clinical data enable systematic identification of patients for genetic testing
Abstract
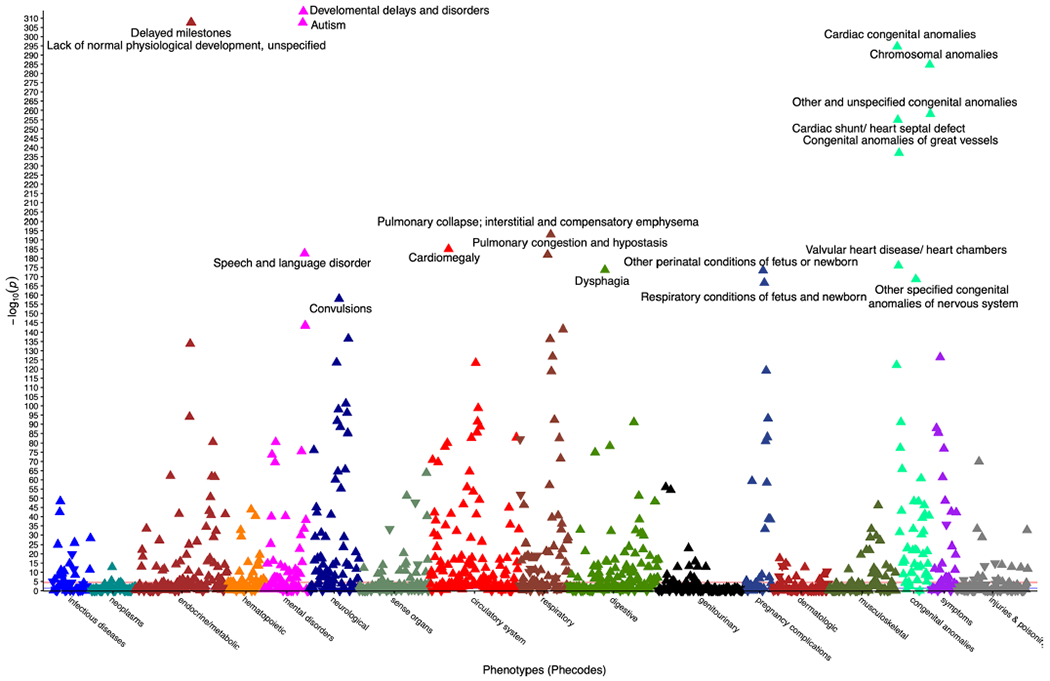
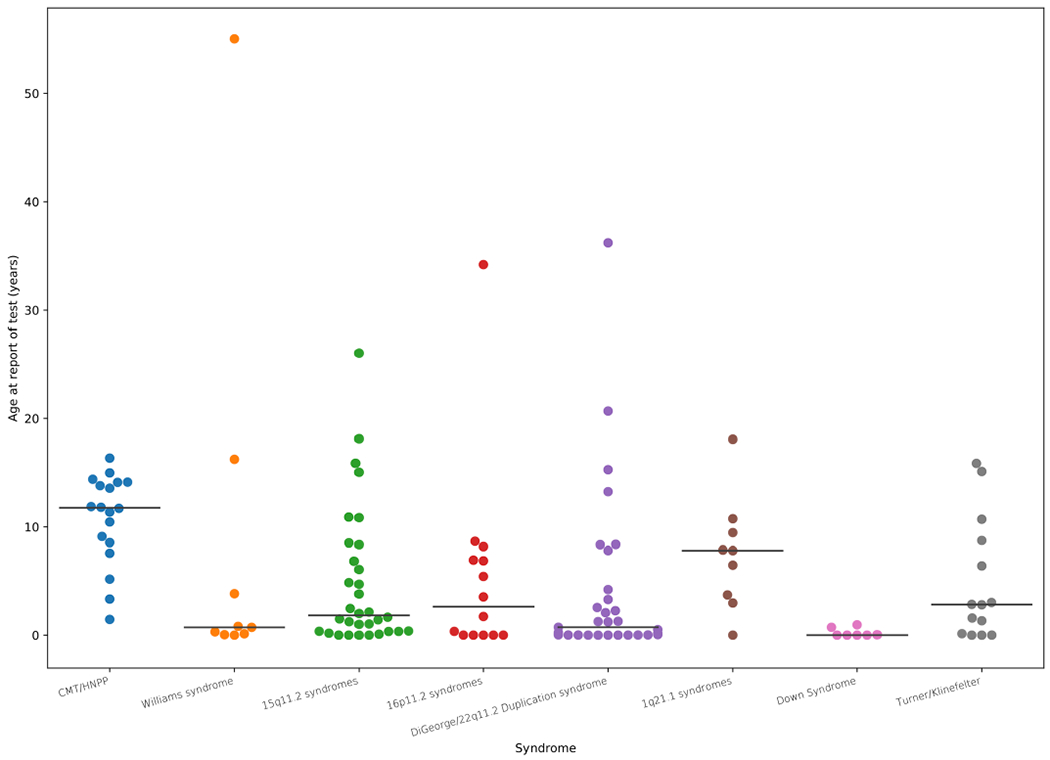
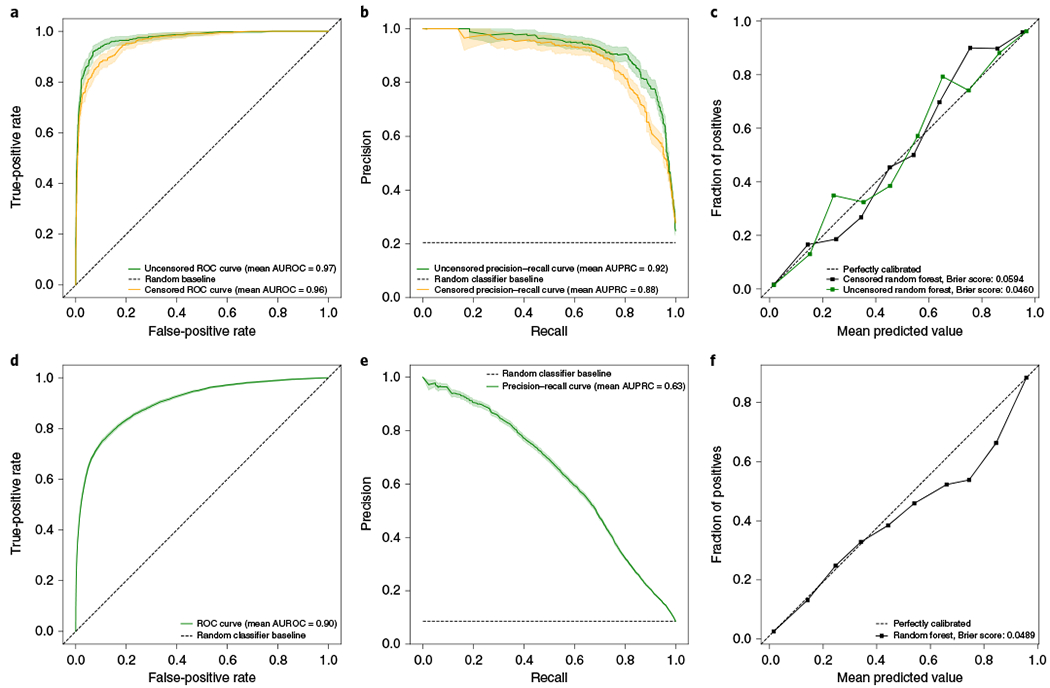
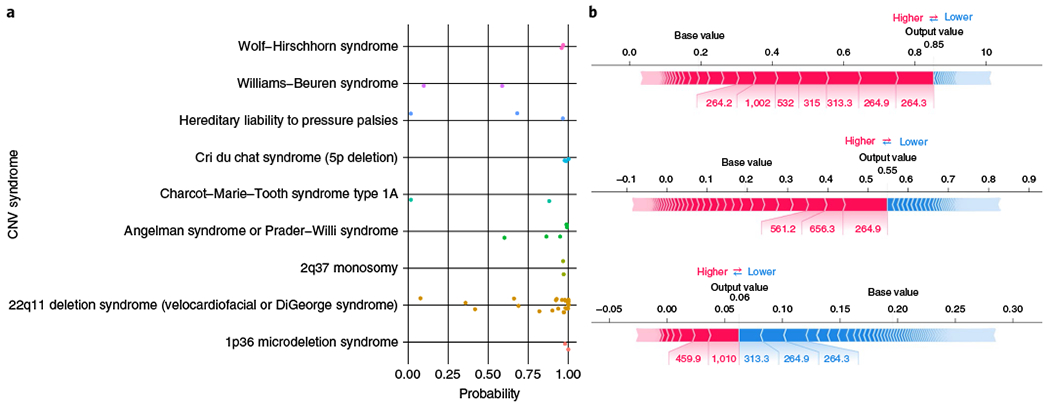
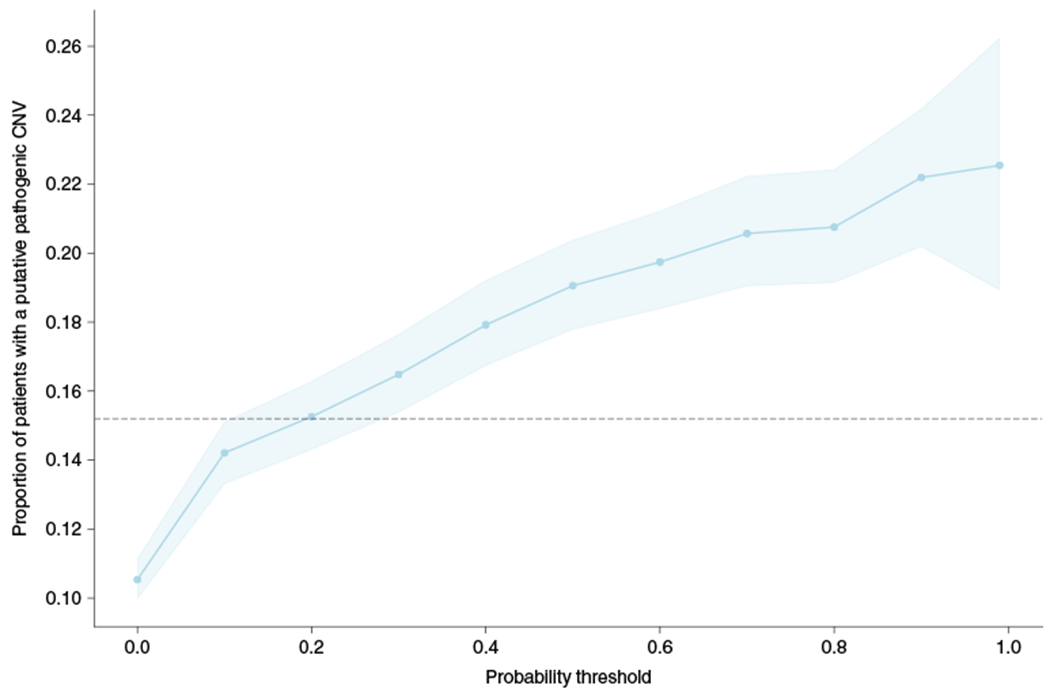
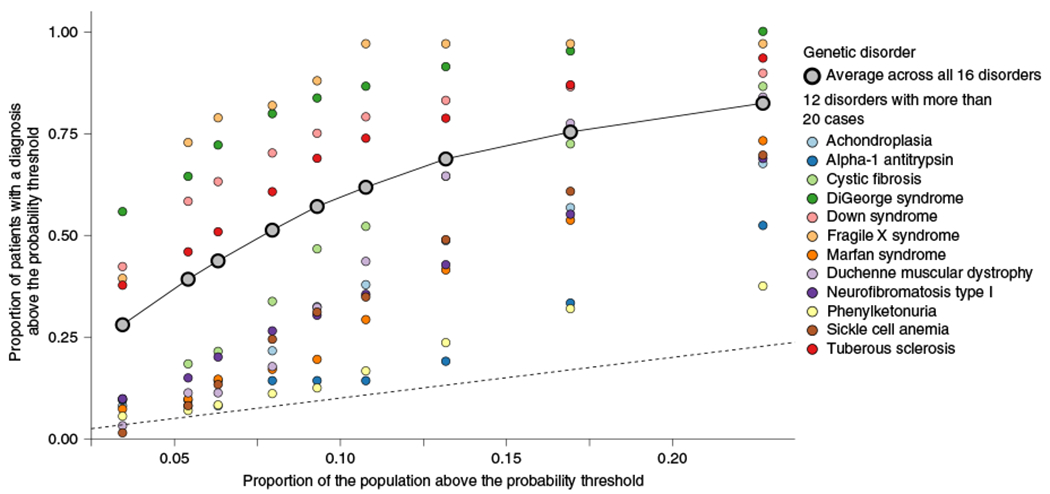
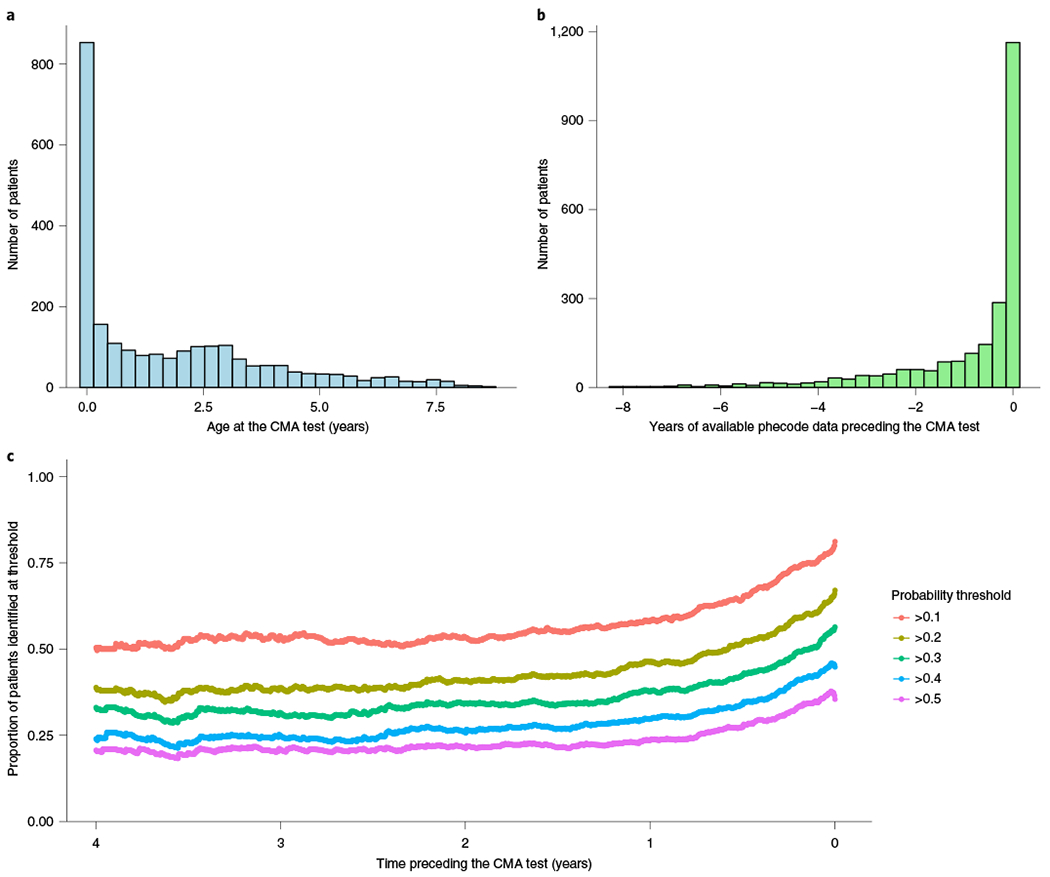
Around 5% of the population is affected by a rare genetic disease, yet most endure years of uncertainty before receiving a genetic test. A common feature of genetic diseases is the presence of multiple rare phenotypes that often span organ systems. Here, we use diagnostic billing information from longitudinal clinical data in the electronic health records (EHRs) of 2,286 patients who received a chromosomal microarray test, and 9,144 matched controls, to build a model to predict who should receive a genetic test. The model achieved high prediction accuracies in a held-out test sample (area under the receiver operating characteristic curve (AUROC), 0.97; area under the precision-recall curve (AUPRC), 0.92), in an independent hospital system (AUROC, 0.95; AUPRC, 0.62), and in an independent set of 172,265 patients in which cases were broadly defined as having an interaction with a genetics provider (AUROC, 0.9; AUPRC, 0.63). Patients carrying a putative pathogenic copy number variant were also accurately identified by the model. Compared with current approaches for genetic test determination, our model could identify more patients for testing while also increasing the proportion of those tested who have a genetic disease. We demonstrate that phenotypic patterns representative of a wide range of genetic diseases can be captured from EHRs to systematize decision-making for genetic testing, with the potential to speed up diagnosis, improve care and reduce costs.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Ferreira CR The burden of rare diseases. Am. J. Med. Genet. A 179, 885–892 (2019). - PubMed
-
- Rosenthal ET, Biesecker LG & Biesecker BB Parental attitudes toward a diagnosis in children with unidentified multiple congenital anomaly syndromes. Am. J. Med. Genet 103, 106–114 (2001). - PubMed
-
- About Rare Diseases (Orphanet, accessed June 2020); https://www.orpha.net/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN
-
- About Rare Diseases (EURORDIS Rare Diseases Europe, accessed June 2020); https://www.eurordis.org/about-rare-diseases
Publication types
MeSH terms
Grants and funding
- UL1 TR000445/TR/NCATS NIH HHS/United States
- U19 HL065962/HL/NHLBI NIH HHS/United States
- R01 LM010685/LM/NLM NIH HHS/United States
- S10 RR025141/RR/NCRR NIH HHS/United States
- R01 HD074711/HD/NICHD NIH HHS/United States
- RC2 GM092618/GM/NIGMS NIH HHS/United States
- P50 GM115305/GM/NIGMS NIH HHS/United States
- R01 MH111776/MH/NIMH NIH HHS/United States
- U01 HG006378/HG/NHGRI NIH HHS/United States
- L30 HG009068/HG/NHGRI NIH HHS/United States
- U01 HG009086/HG/NHGRI NIH HHS/United States
- R01 MH113362/MH/NIMH NIH HHS/United States
- UL1 RR024975/RR/NCRR NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 NS032830/NS/NINDS NIH HHS/United States
- U01 HG004798/HG/NHGRI NIH HHS/United States
- UL1 TR002243/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
