

A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance
- PMID: 34085047
- PMCID: PMC8171279
- DOI: 10.1038/s43018-020-00125-0
A MYC and RAS co-activation signature in localized prostate cancer drives bone metastasis and castration resistance
Abstract
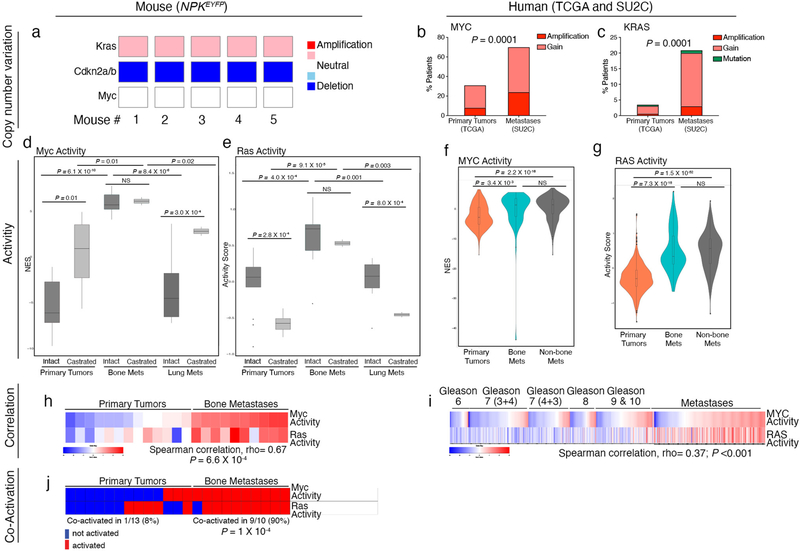
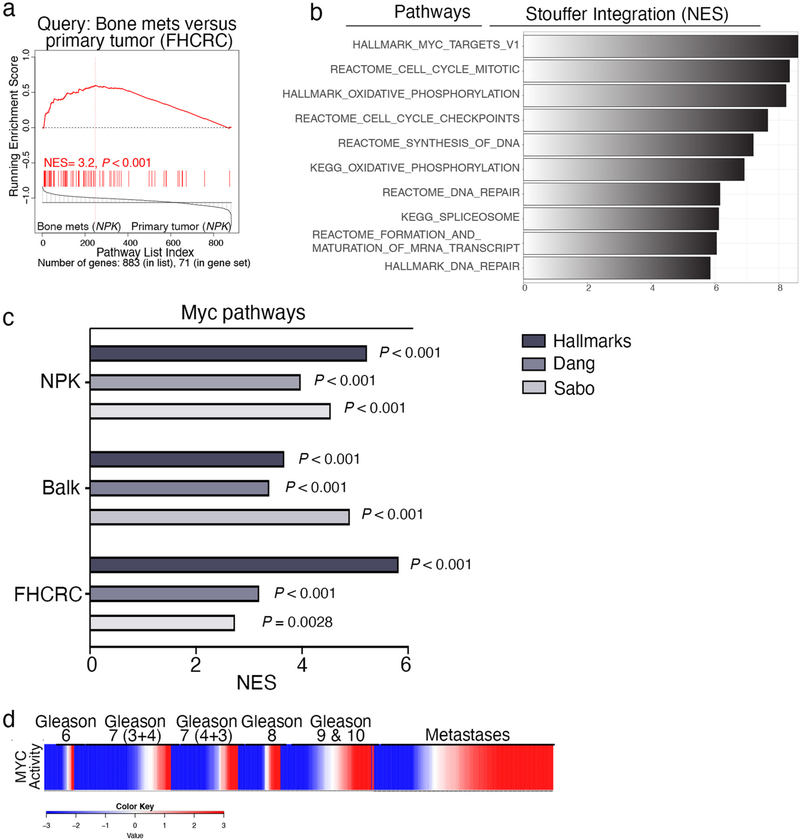
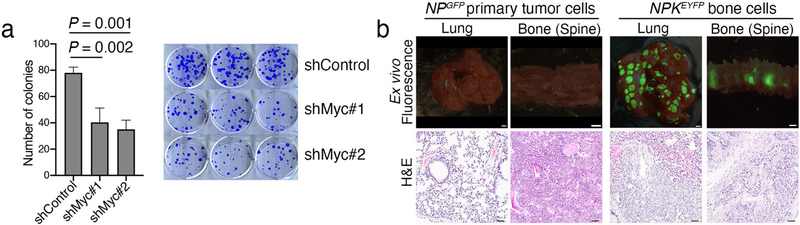
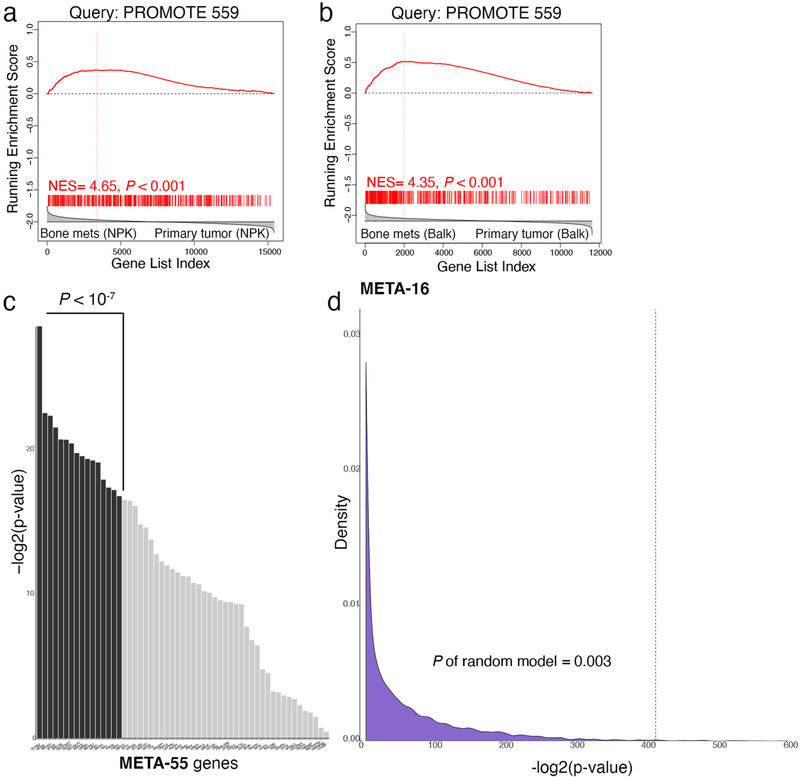
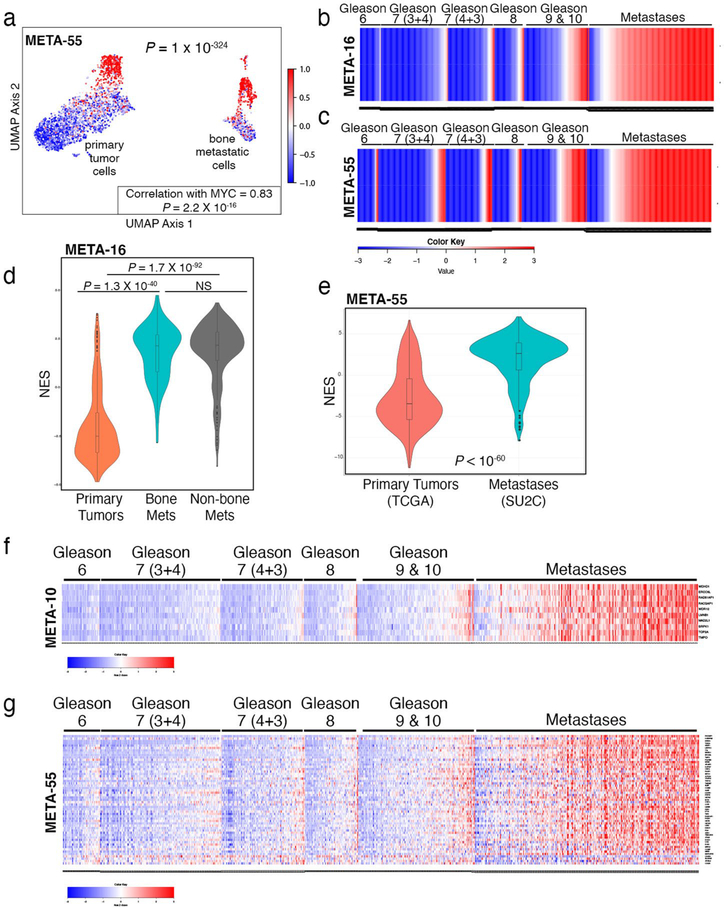
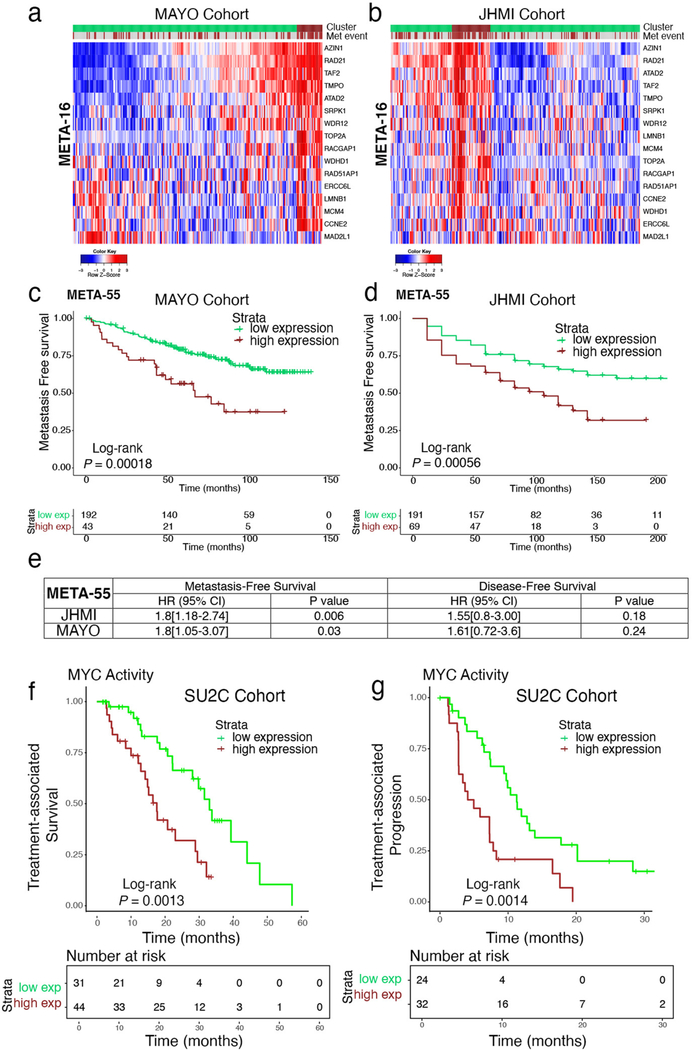
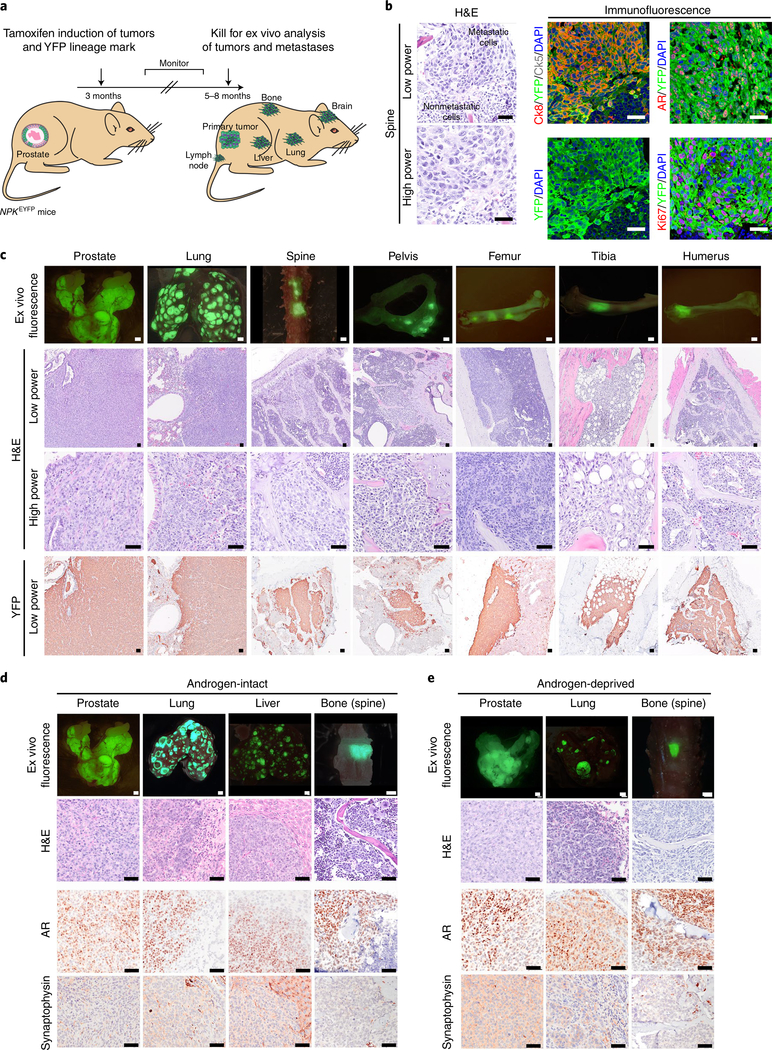
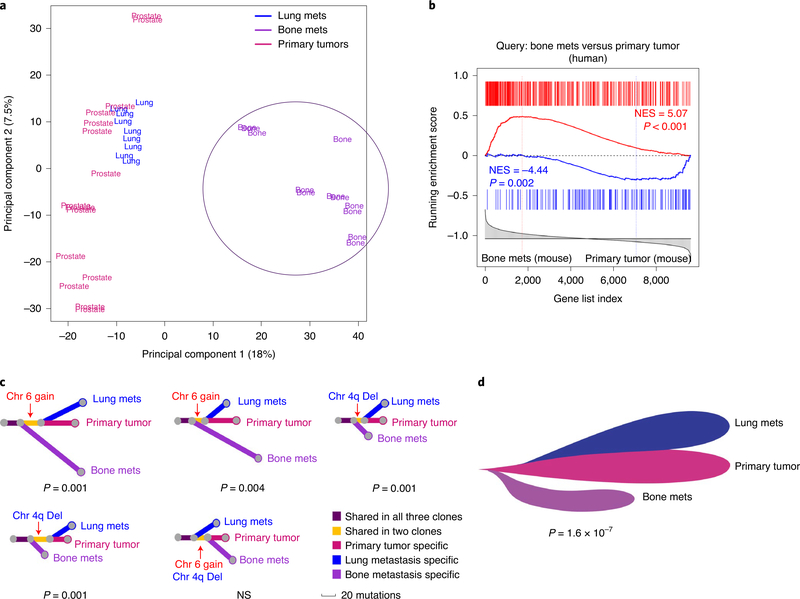
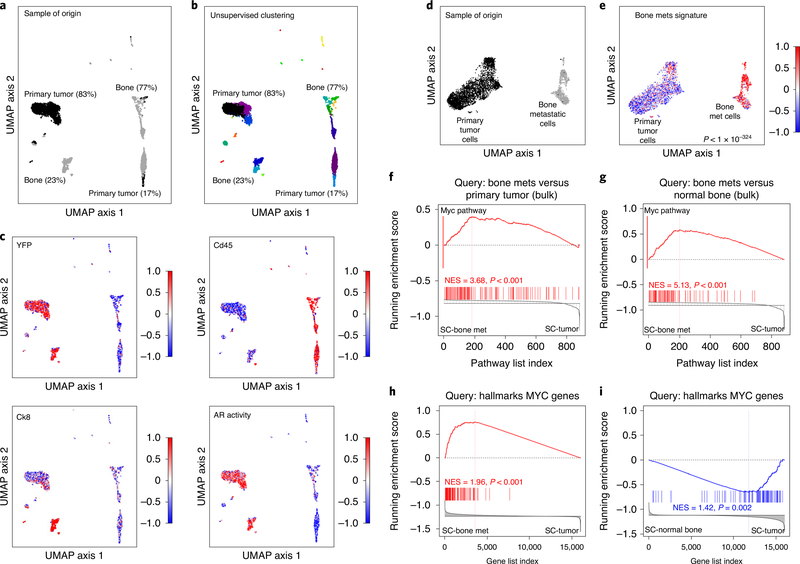
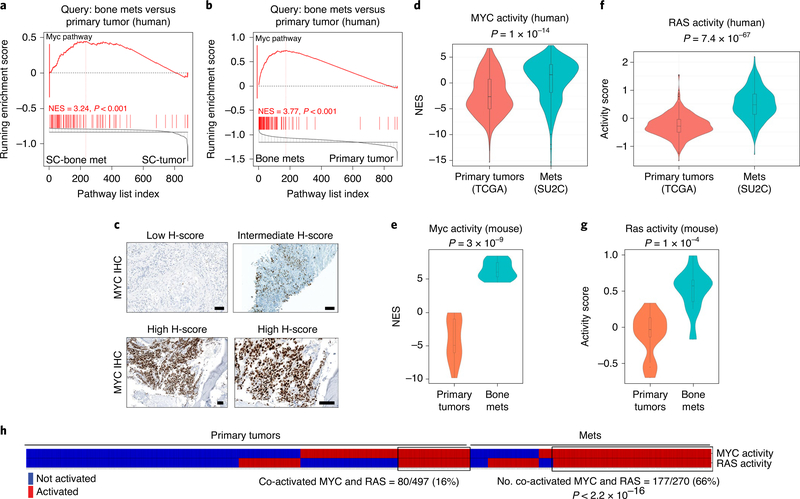
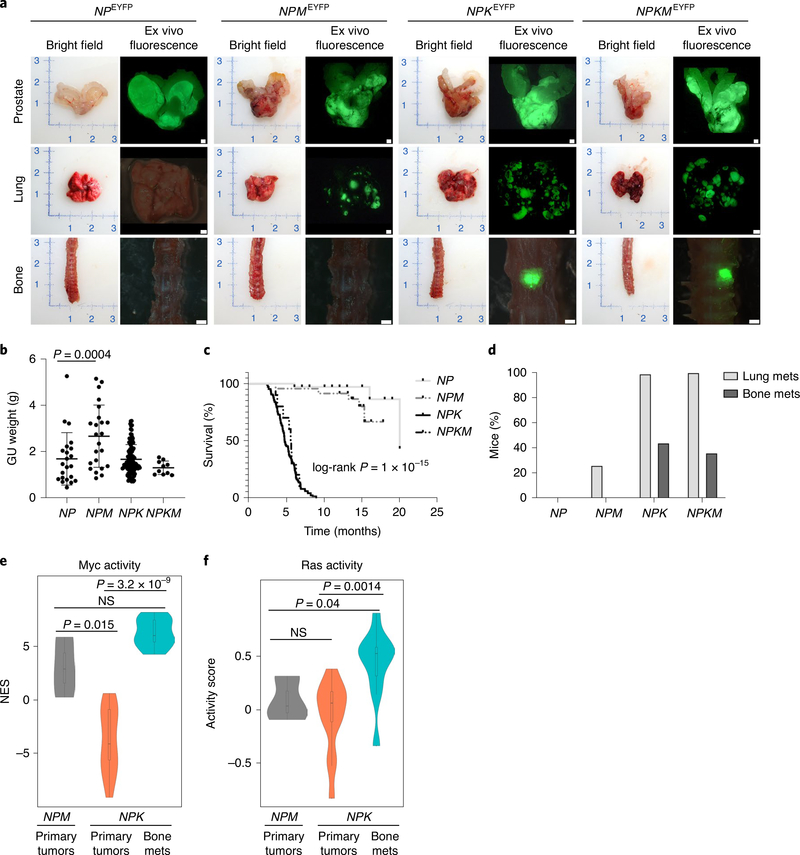
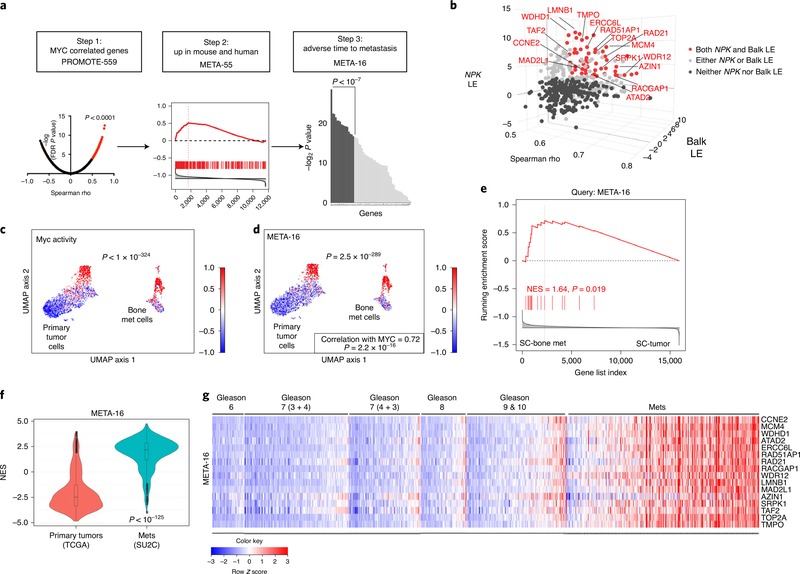
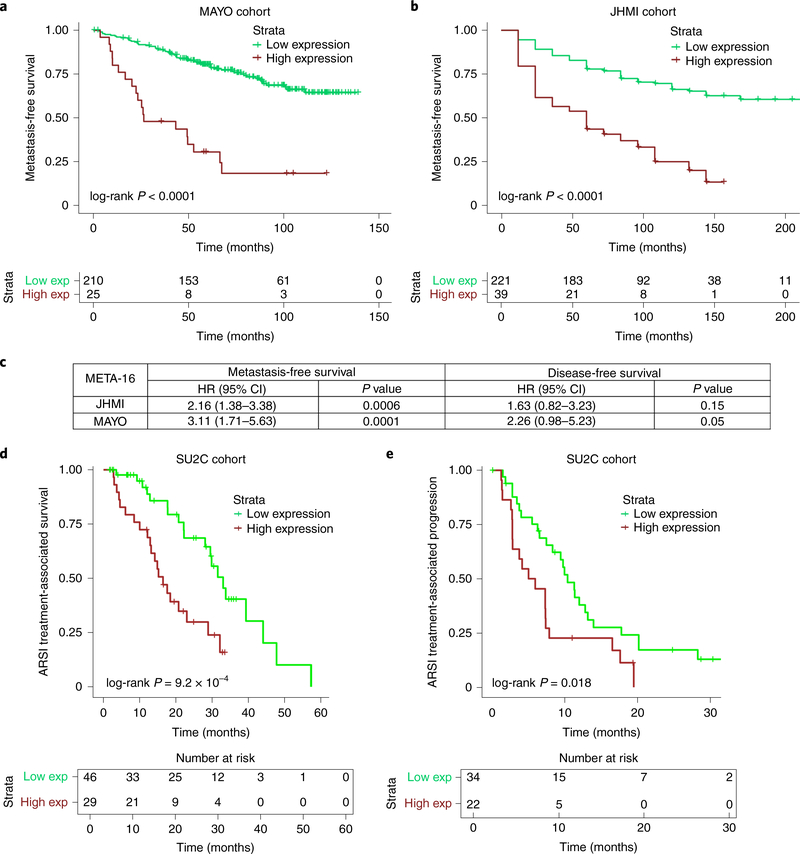
Understanding the intricacies of lethal prostate cancer poses specific challenges due to difficulties in accurate modeling of metastasis in vivo. Here we show that NPK EYFP mice (for Nkx3.1 CreERT2/+ ; Pten flox/flox ; Kras LSL-G12D/+ ; R26R-CAG-LSL-EYFP/+) develop prostate cancer with a high penetrance of metastasis to bone, thereby enabling detection and tracking of bone metastasis in vivo and ex vivo. Transcriptomic and whole-exome analyses of bone metastasis from these mice revealed distinct molecular profiles conserved between human and mouse and specific patterns of subclonal branching from the primary tumor. Integrating bulk and single-cell transcriptomic data from mouse and human datasets with functional studies in vivo unravels a unique MYC/RAS co-activation signature associated with prostate cancer metastasis. Finally, we identify a gene signature with prognostic value for time to metastasis and predictive of treatment response in human patients undergoing androgen receptor therapy across clinical cohorts, thus uncovering conserved mechanisms of metastasis with potential translational significance.
Figures





References
-
- Sartor O & de Bono JS Metastatic prostate cancer. N. Engl. J. Med 378, 645–657 (2018). - PubMed
-
- Scher HI & Sawyers CL Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol 23, 8253–8261 (2005). - PubMed
-
- Rickman DS, Beltran H, Demichelis F & Rubin MA Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med 23, 1–10 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA244780/CA/NCI NIH HHS/United States
- U54 CA209997/CA/NCI NIH HHS/United States
- R01 CA218024/CA/NCI NIH HHS/United States
- P30 CA006973/CA/NCI NIH HHS/United States
- R01 CA109182/CA/NCI NIH HHS/United States
- P30 CA013696/CA/NCI NIH HHS/United States
- R01 CA193442/CA/NCI NIH HHS/United States
- P50 CA211024/CA/NCI NIH HHS/United States
- UL1 TR001873/TR/NCATS NIH HHS/United States
- F31 CA210607/CA/NCI NIH HHS/United States
- R01 CA183929/CA/NCI NIH HHS/United States
- P50 CA097186/CA/NCI NIH HHS/United States
- R01 CA173481/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
