

Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma
- PMID: 34085048
- PMCID: PMC8168620
- DOI: 10.1038/s43018-020-00160-x
Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma
Abstract
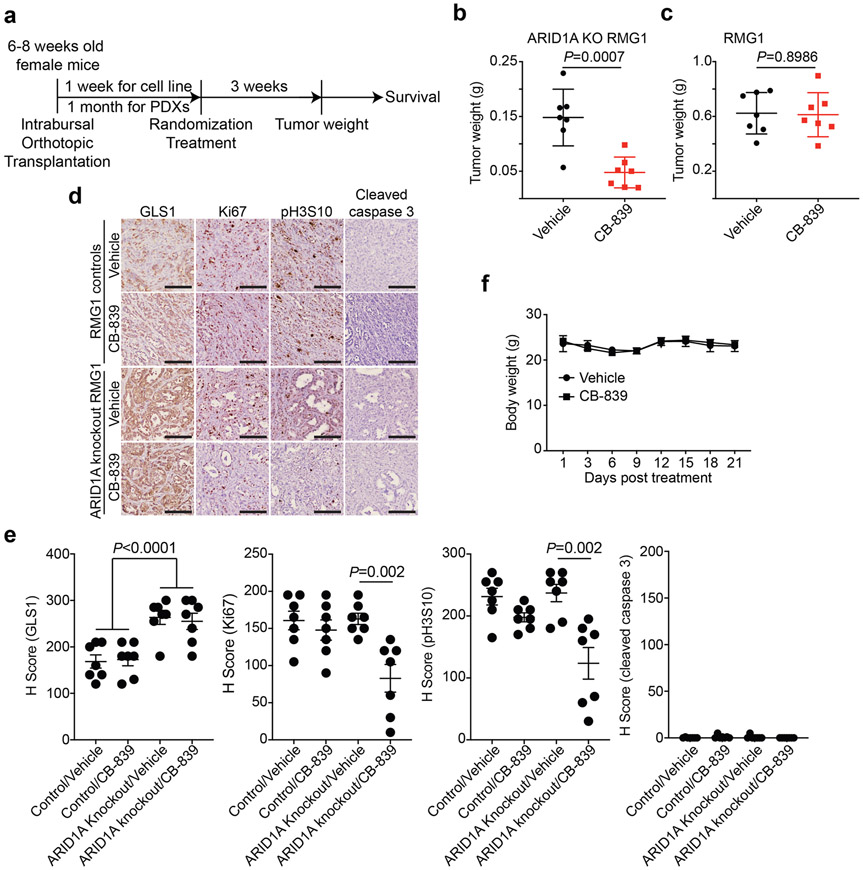
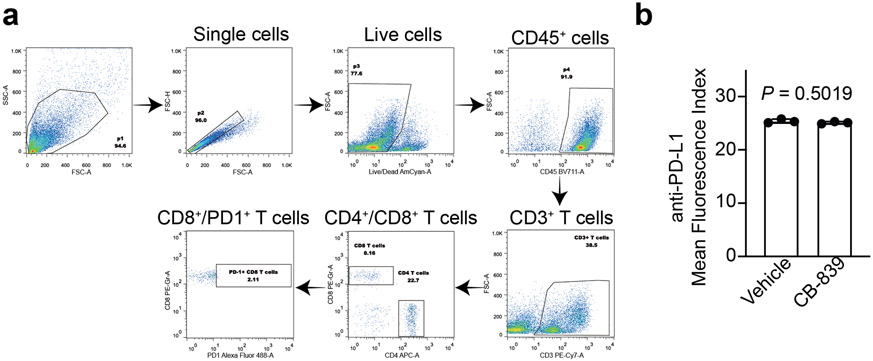
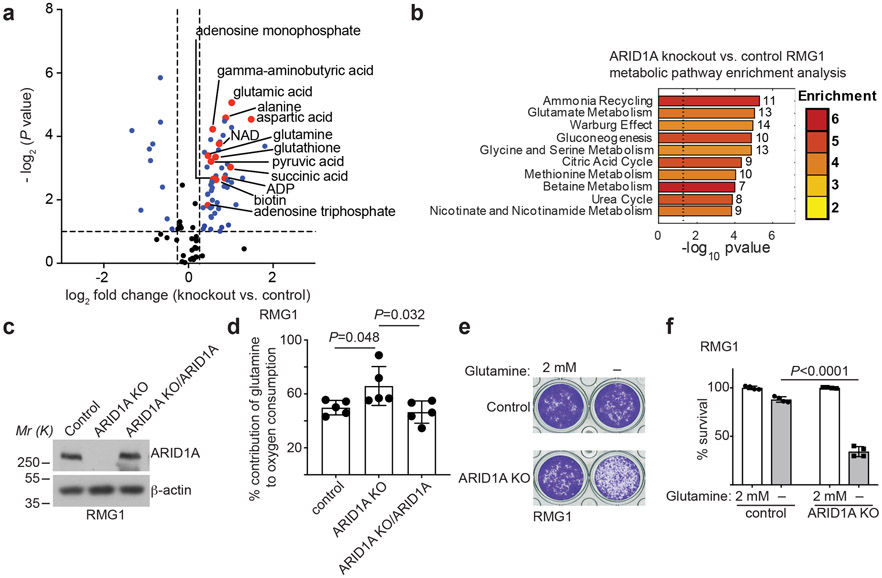
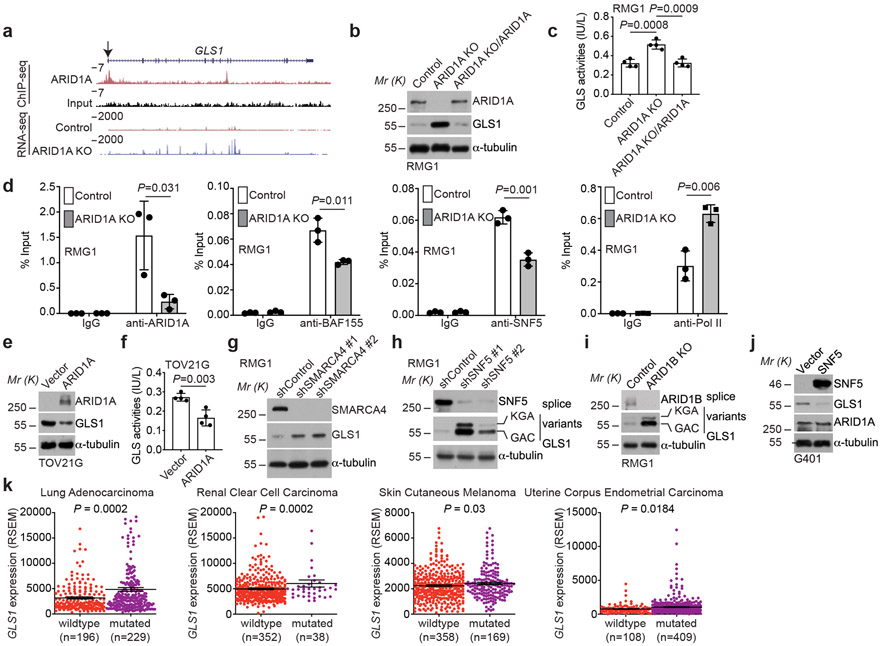
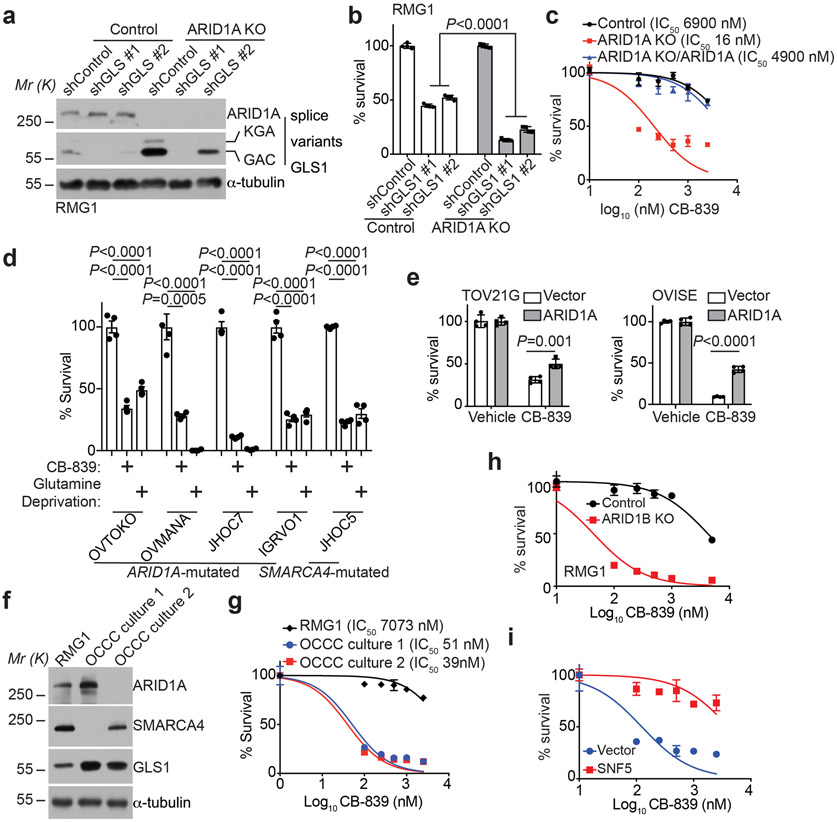
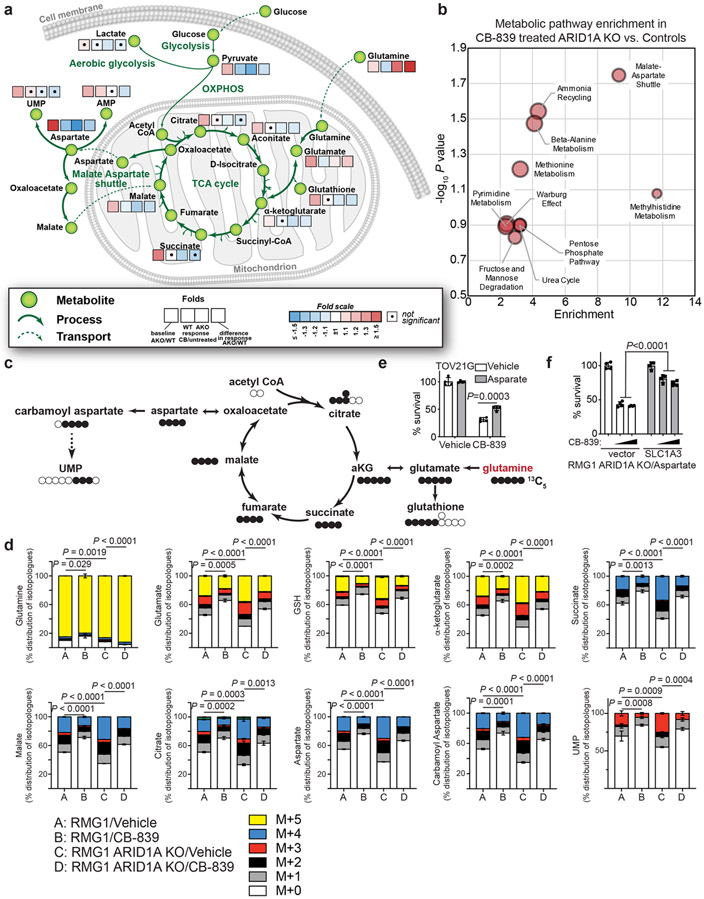
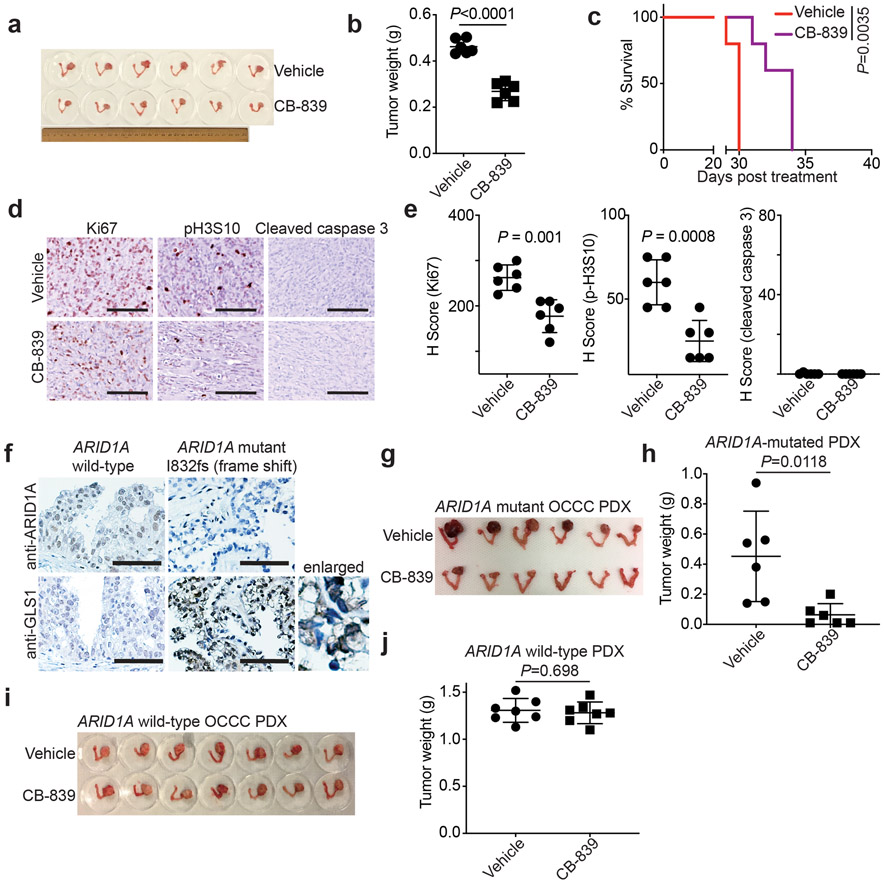
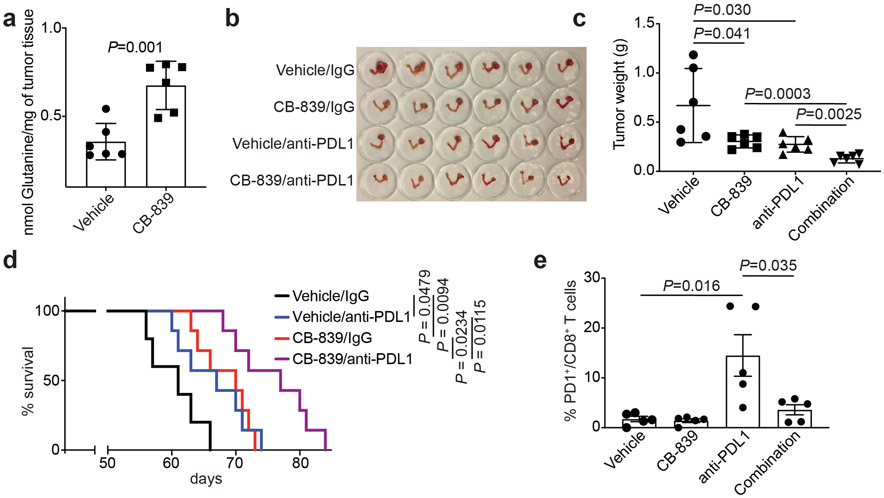
Alterations in components of the SWI/SNF chromatin-remodeling complex occur in ~20% of all human cancers. For example, ARID1A is mutated in up to 62% of clear cell ovarian carcinoma (OCCC), a disease currently lacking effective therapies. Here we show that ARID1A mutation creates a dependence on glutamine metabolism. SWI/SNF represses glutaminase (GLS1) and ARID1A inactivation upregulates GLS1. ARID1A inactivation increases glutamine utilization and metabolism through the tricarboxylic acid cycle to support aspartate synthesis. Indeed, glutaminase inhibitor CB-839 suppresses the growth of ARID1A mutant, but not wildtype, OCCCs in both orthotopic and patient-derived xenografts. In addition, glutaminase inhibitor CB-839 synergizes with immune checkpoint blockade anti-PDL1 antibody in a genetic OCCC mouse model driven by conditional Arid1a inactivation. Our data indicate that pharmacological inhibition of glutaminase alone or in combination with immune checkpoint blockade represents an effective therapeutic strategy for cancers involving alterations in the SWI/SNF complex such as ARID1A mutations.
Figures
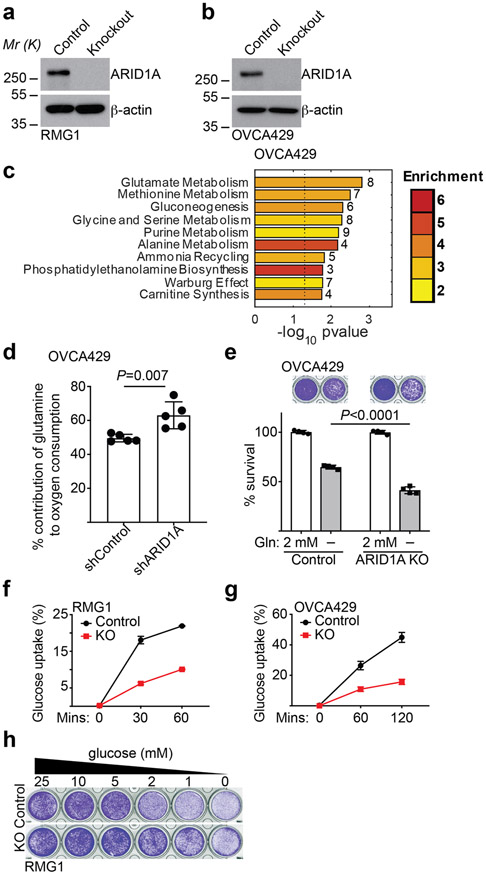
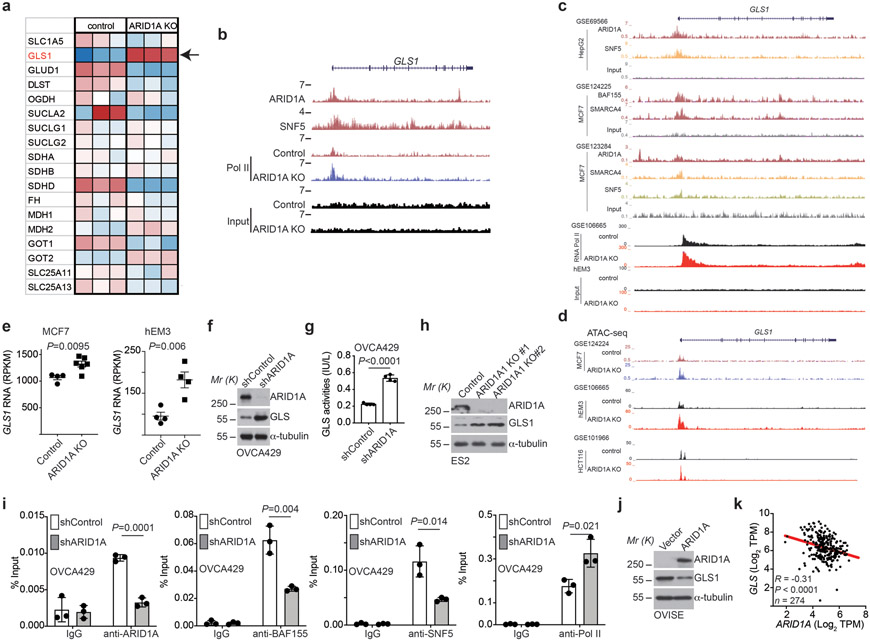
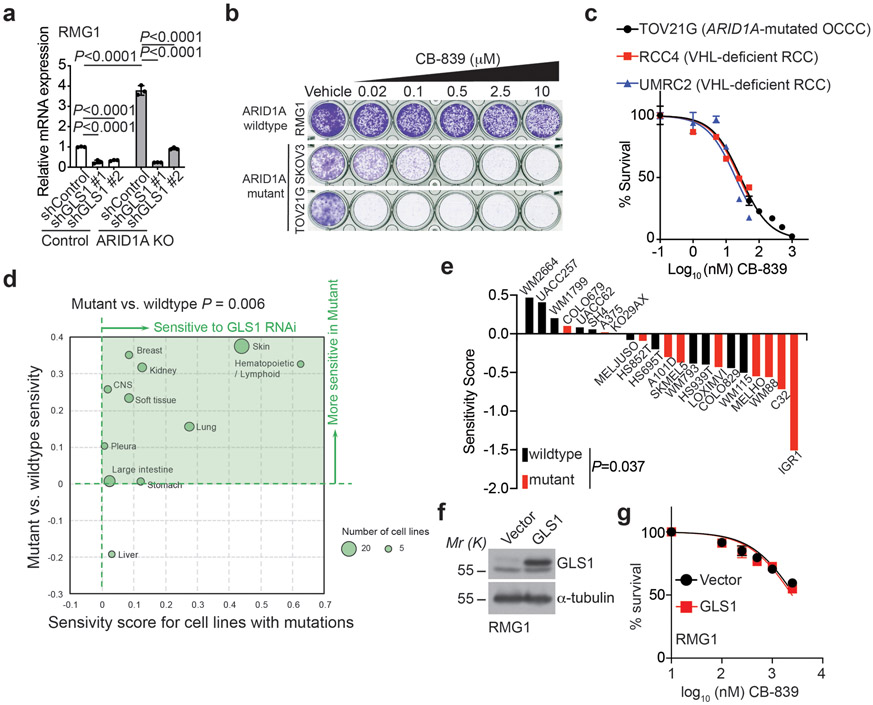
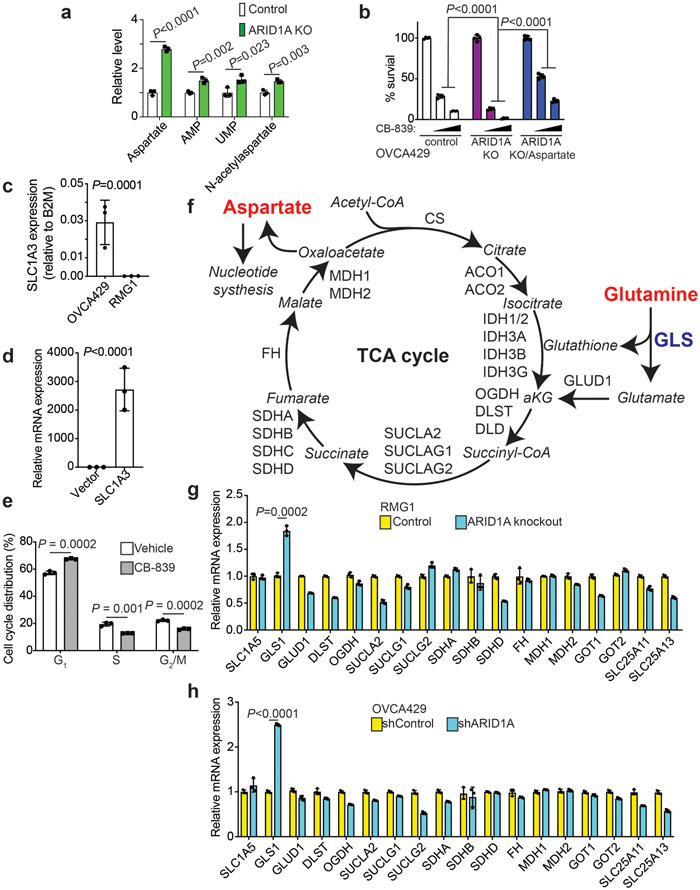
References
-
- Wilson BG & Roberts CW SWI/SNF nucleosome remodellers and cancer. Nature reviews. Cancer 11, 481–492 (2011). - PubMed
Methods-only References
-
- Skene PJ, Henikoff JG & Henikoff S Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13, 1006–1019 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K99 CA241395/CA/NCI NIH HHS/United States
- S10 OD023586/OD/NIH HHS/United States
- P01 CA114046/CA/NCI NIH HHS/United States
- P01 AG031862/AG/NIA NIH HHS/United States
- P50 CA228991/CA/NCI NIH HHS/United States
- R50 CA211199/CA/NCI NIH HHS/United States
- R01 CA202919/CA/NCI NIH HHS/United States
- R01 CA239128/CA/NCI NIH HHS/United States
- F31 CA247336/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R50 CA221838/CA/NCI NIH HHS/United States
- R00 CA241395/CA/NCI NIH HHS/United States
- R01 CA195670/CA/NCI NIH HHS/United States
- R01 CA160331/CA/NCI NIH HHS/United States
- R01 CA163377/CA/NCI NIH HHS/United States
- S10 OD023658/OD/NIH HHS/United States
- DP2 CA249950/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
