

Spatially resolved free-energy contributions of native fold and molten-globule-like Crambin
- PMID: 34087209
- PMCID: PMC8391029
- DOI: 10.1016/j.bpj.2021.05.019
Spatially resolved free-energy contributions of native fold and molten-globule-like Crambin
Abstract
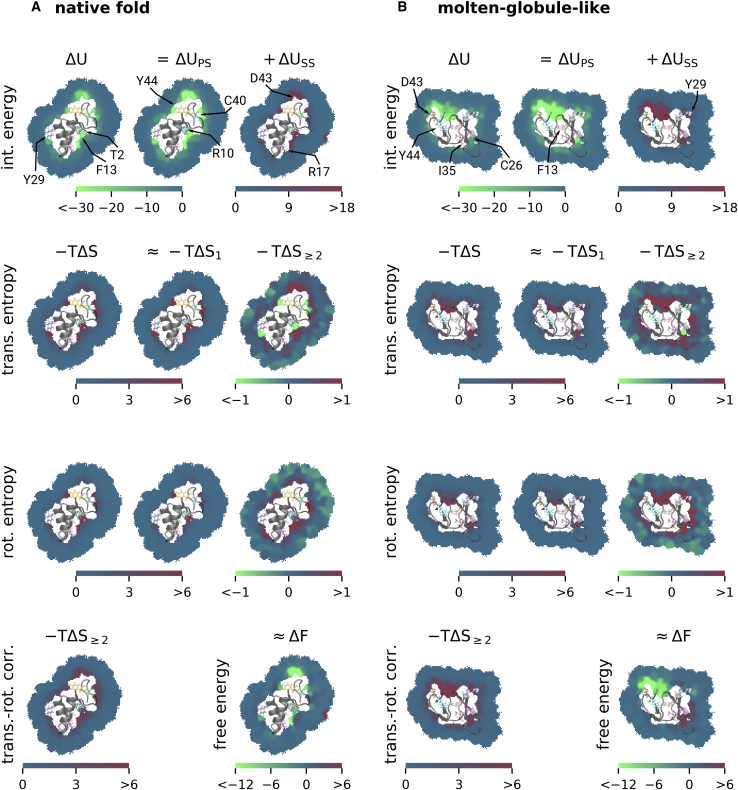
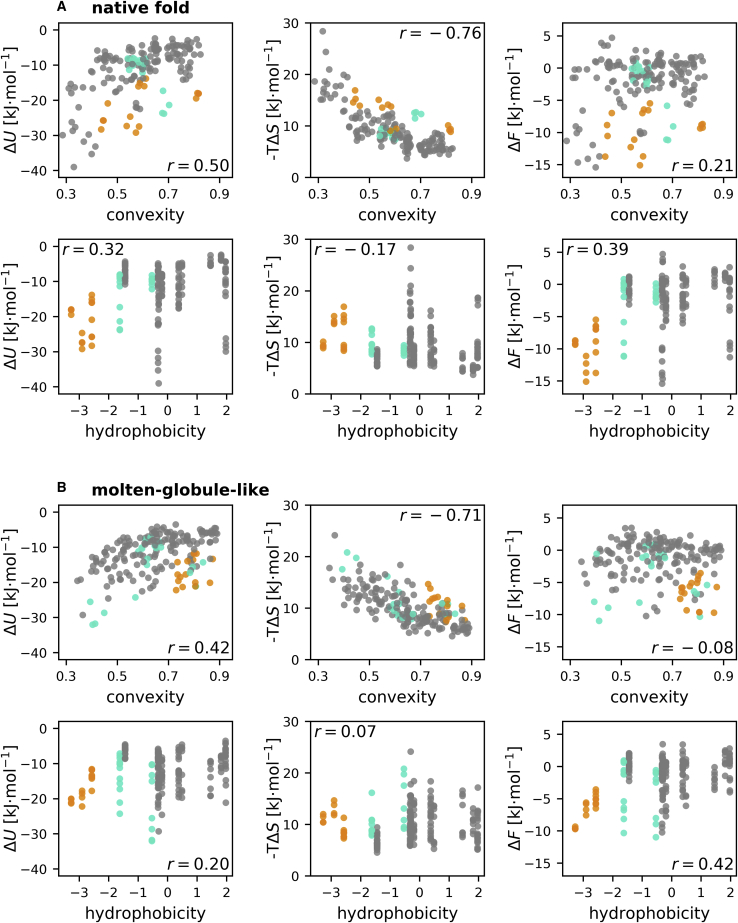
The folding stability of a protein is governed by the free-energy difference between its folded and unfolded states, which results from a delicate balance of much larger but almost compensating enthalpic and entropic contributions. The balance can therefore easily be shifted by an external disturbance, such as a mutation of a single amino acid or a change of temperature, in which case the protein unfolds. Effects such as cold denaturation, in which a protein unfolds because of cooling, provide evidence that proteins are strongly stabilized by the solvent entropy contribution to the free-energy balance. However, the molecular mechanisms behind this solvent-driven stability, their quantitative contribution in relation to other free-energy contributions, and how the involved solvent thermodynamics is affected by individual amino acids are largely unclear. Therefore, we addressed these questions using atomistic molecular dynamics simulations of the small protein Crambin in its native fold and a molten-globule-like conformation, which here served as a model for the unfolded state. The free-energy difference between these conformations was decomposed into enthalpic and entropic contributions from the protein and spatially resolved solvent contributions using the nonparametric method Per|Mut. From the spatial resolution, we quantified the local effects on the solvent free-energy difference at each amino acid and identified dependencies of the local enthalpy and entropy on the protein curvature. We identified a strong stabilization of the native fold by almost 500 kJ mol-1 due to the solvent entropy, revealing it as an essential contribution to the total free-energy difference of (53 ± 84) kJ mol-1. Remarkably, more than half of the solvent entropy contribution arose from induced water correlations.
Copyright © 2021 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Privalov P.L., Khechinashvili N.N. A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J. Mol. Biol. 1974;86:665–684. - PubMed
-
- Dias C.L., Ala-Nissila T., Karttunen M. The hydrophobic effect and its role in cold denaturation. Cryobiology. 2010;60:91–99. - PubMed
-
- Agashe V.R., Udgaonkar J.B. Thermodynamics of denaturation of barstar: evidence for cold denaturation and evaluation of the interaction with guanidine hydrochloride. Biochemistry. 1995;34:3286–3299. - PubMed
-
- Wendler K., Thar J., Kirchner B. Estimating the hydrogen bond energy. J. Phys. Chem. A. 2010;114:9529–9536. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
