

Long non-coding RNA exploration for mesenchymal stem cell characterisation
- PMID: 34088266
- PMCID: PMC8178833
- DOI: 10.1186/s12864-020-07289-0
Long non-coding RNA exploration for mesenchymal stem cell characterisation
Abstract
Background: The development of RNA sequencing (RNAseq) and the corresponding emergence of public datasets have created new avenues of transcriptional marker search. The long non-coding RNAs (lncRNAs) constitute an emerging class of transcripts with a potential for high tissue specificity and function. Therefore, we tested the biomarker potential of lncRNAs on Mesenchymal Stem Cells (MSCs), a complex type of adult multipotent stem cells of diverse tissue origins, that is frequently used in clinics but which is lacking extensive characterization.
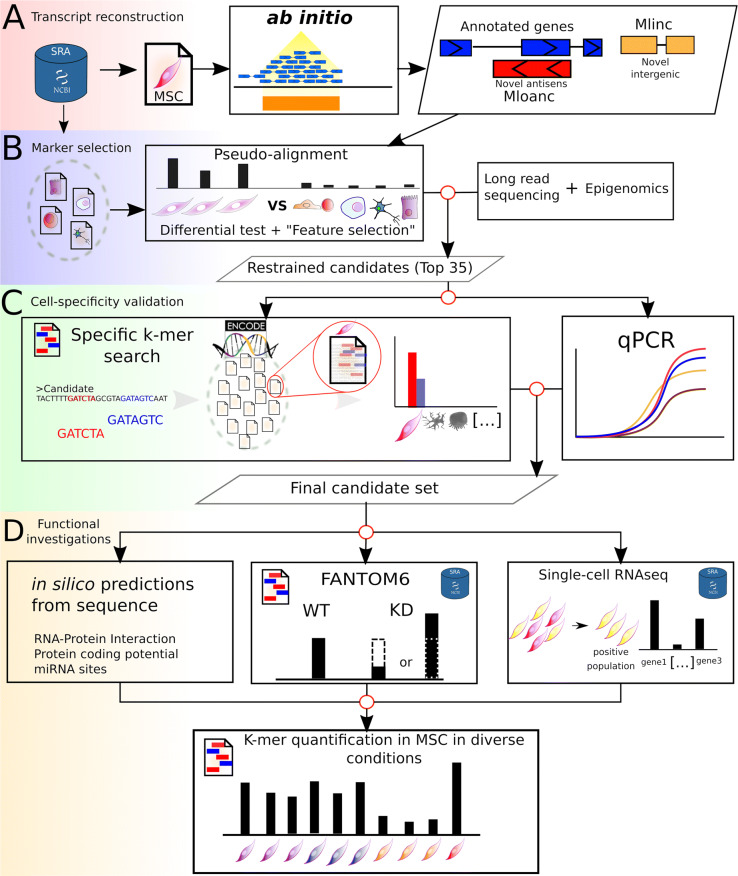
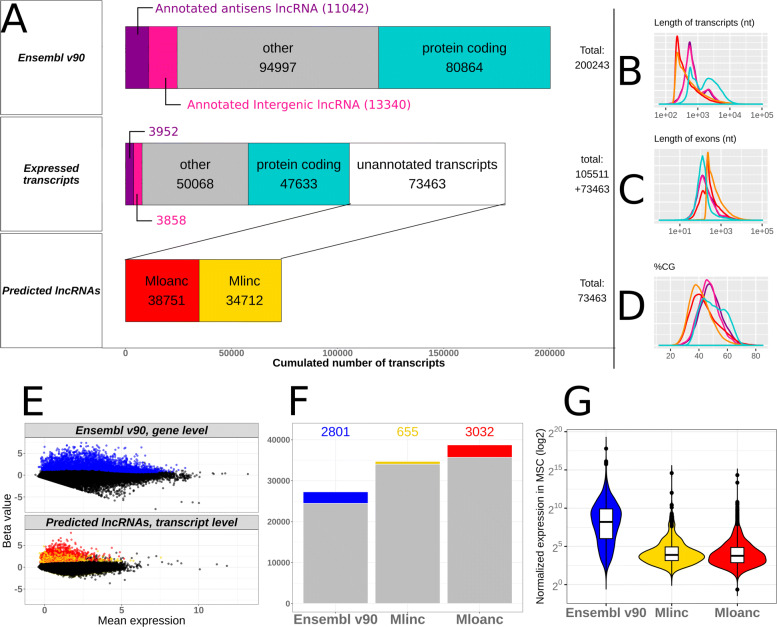
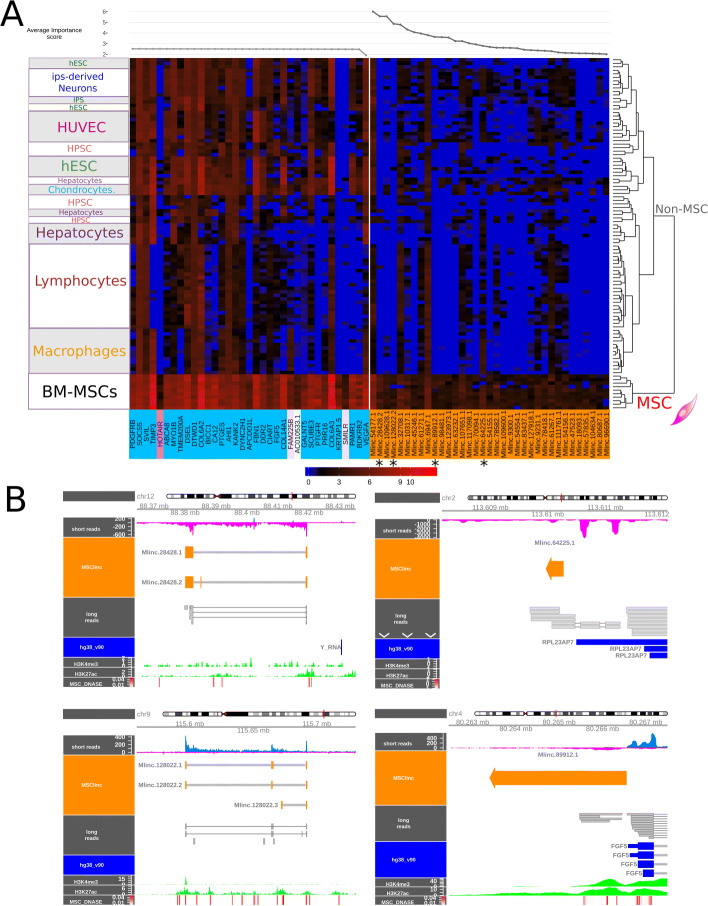
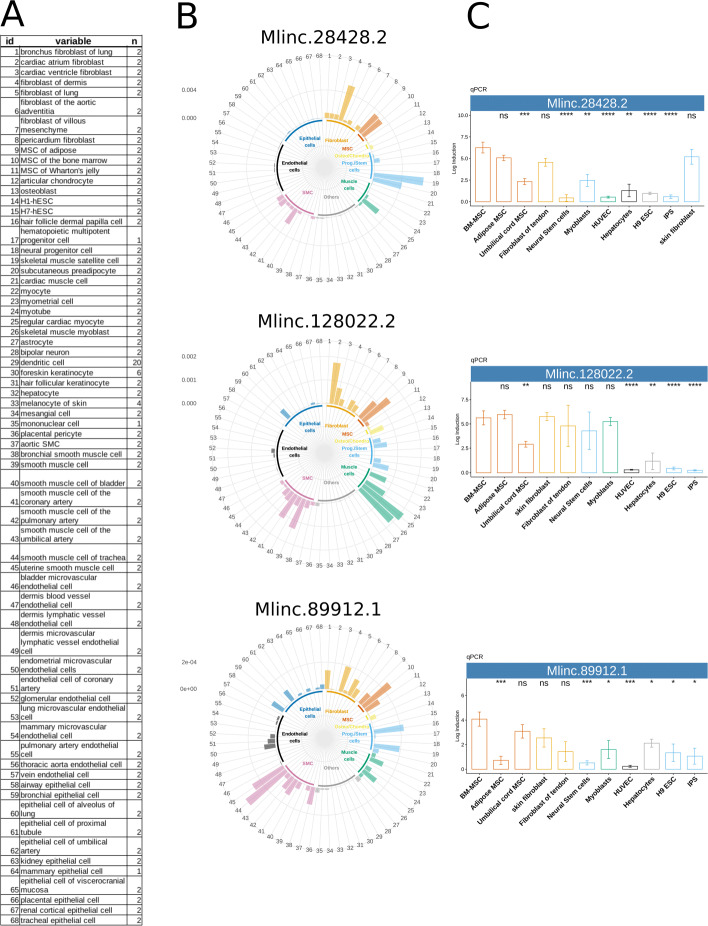
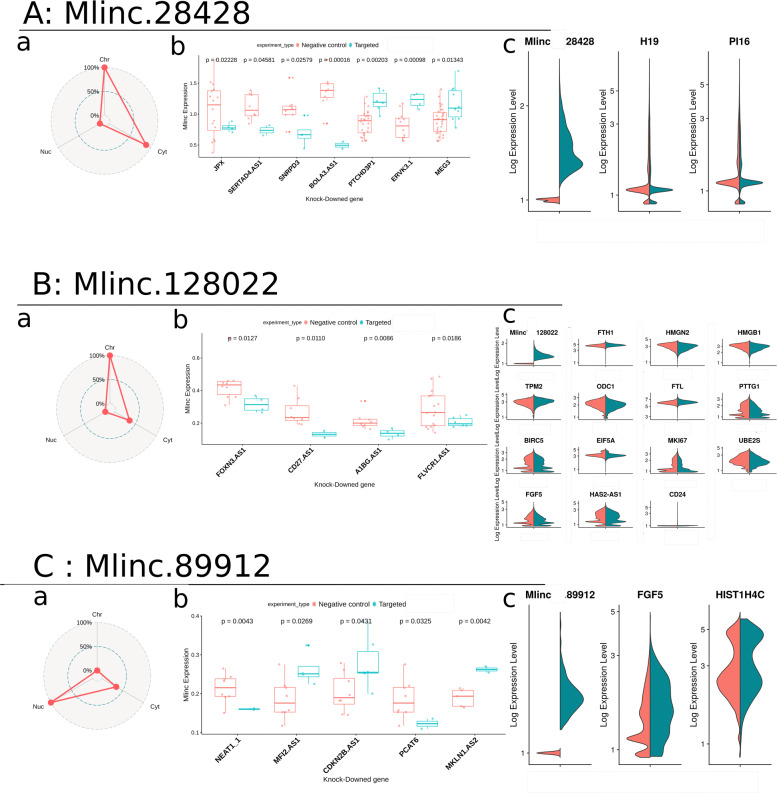
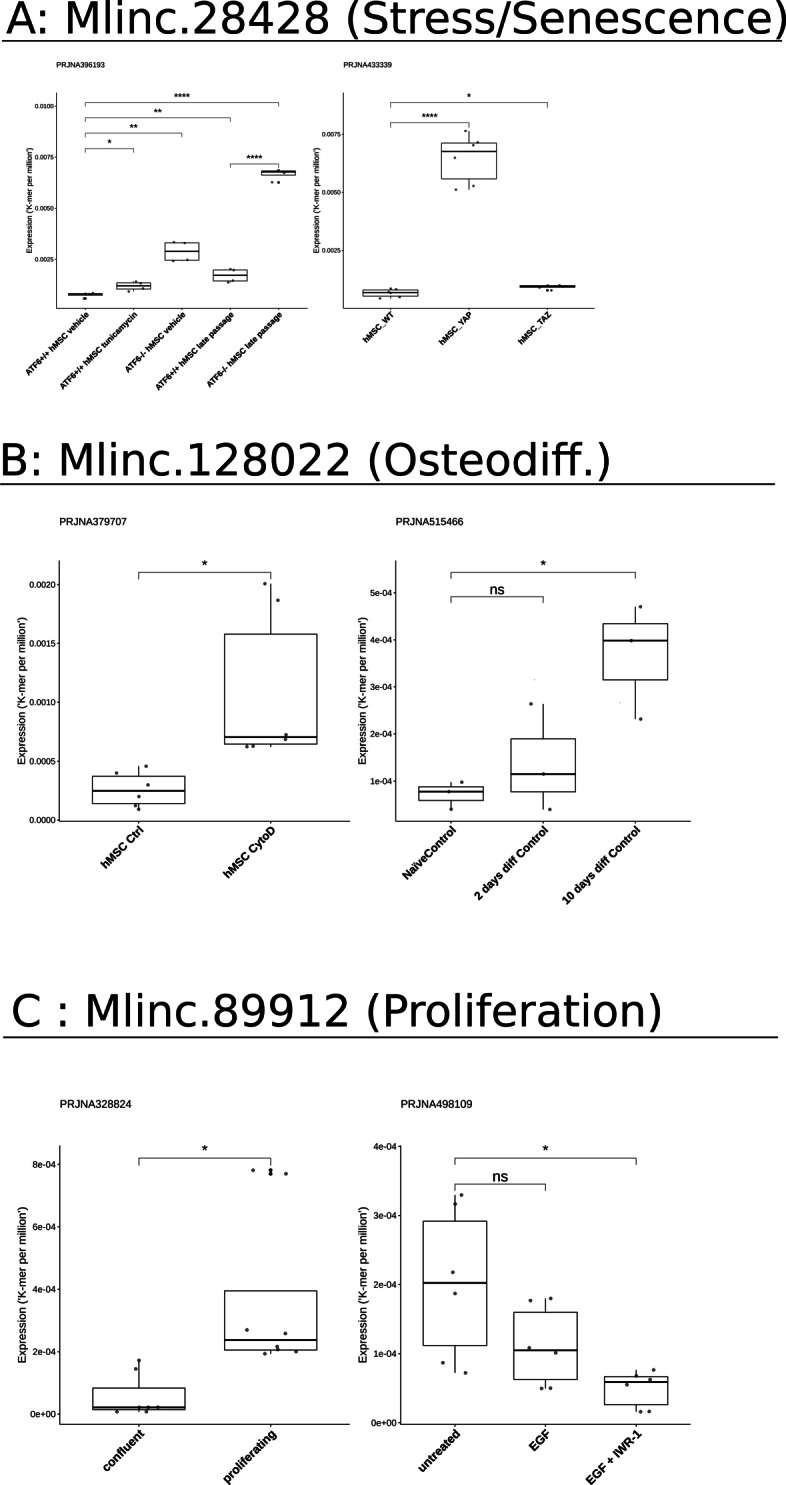
Results: We developed a dedicated bioinformatics pipeline for the purpose of building a cell-specific catalogue of unannotated lncRNAs. The pipeline performs ab initio transcript identification, pseudoalignment and uses new methodologies such as a specific k-mer approach for naive quantification of expression in numerous RNAseq data. We next applied it on MSCs, and our pipeline was able to highlight novel lncRNAs with high cell specificity. Furthermore, with original and efficient approaches for functional prediction, we demonstrated that each candidate represents one specific state of MSCs biology.
Conclusions: We showed that our approach can be employed to harness lncRNAs as cell markers. More specifically, our results suggest different candidates as potential actors in MSCs biology and propose promising directions for future experimental investigations.
Keywords: Bioinformatics; Long non-coding RNA; Mesenchymal stem cell; NGS analysis; RNAseq; Transcriptomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
