

Ultra-accurate microbial amplicon sequencing with synthetic long reads
- PMID: 34090540
- PMCID: PMC8179091
- DOI: 10.1186/s40168-021-01072-3
Ultra-accurate microbial amplicon sequencing with synthetic long reads
Abstract
Background: Out of the many pathogenic bacterial species that are known, only a fraction are readily identifiable directly from a complex microbial community using standard next generation DNA sequencing. Long-read sequencing offers the potential to identify a wider range of species and to differentiate between strains within a species, but attaining sufficient accuracy in complex metagenomes remains a challenge.
Methods: Here, we describe and analytically validate LoopSeq, a commercially available synthetic long-read (SLR) sequencing technology that generates highly accurate long reads from standard short reads.
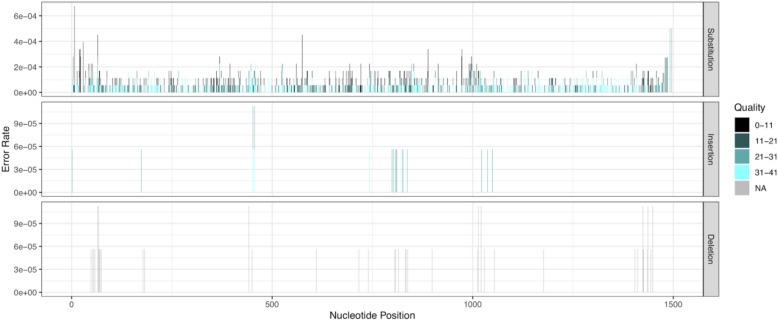
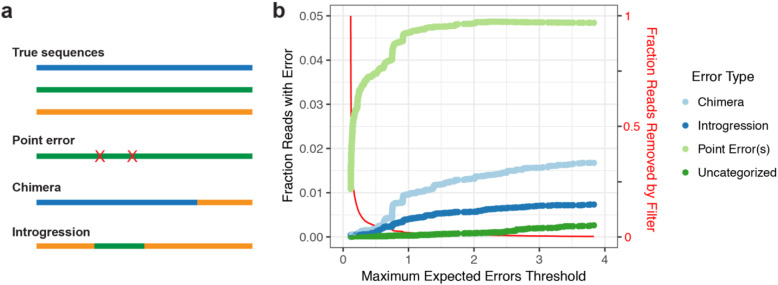
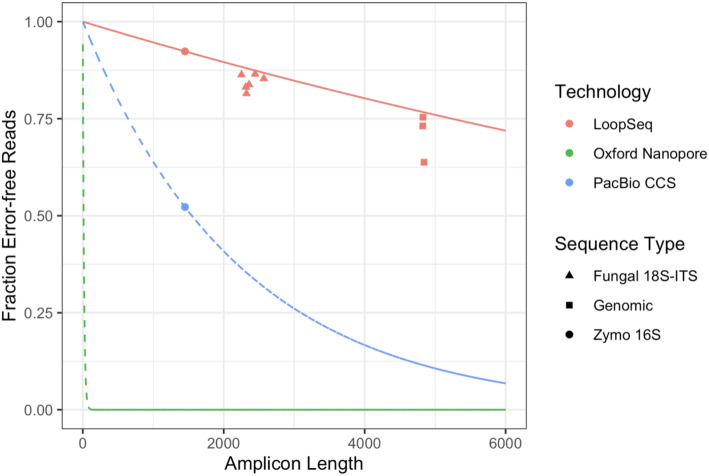
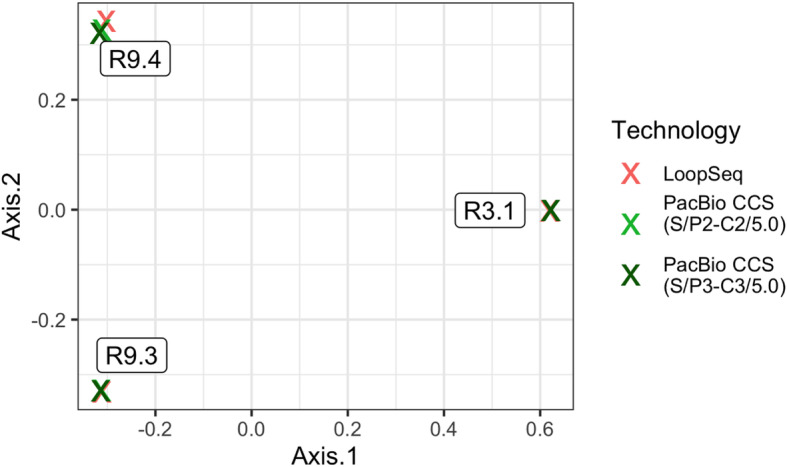
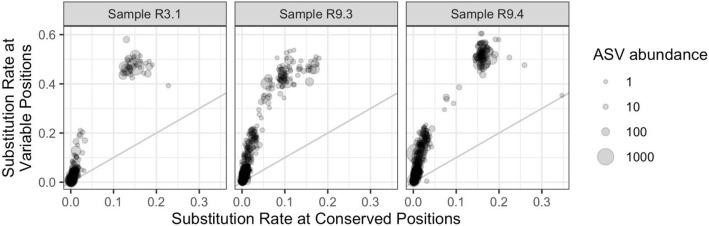
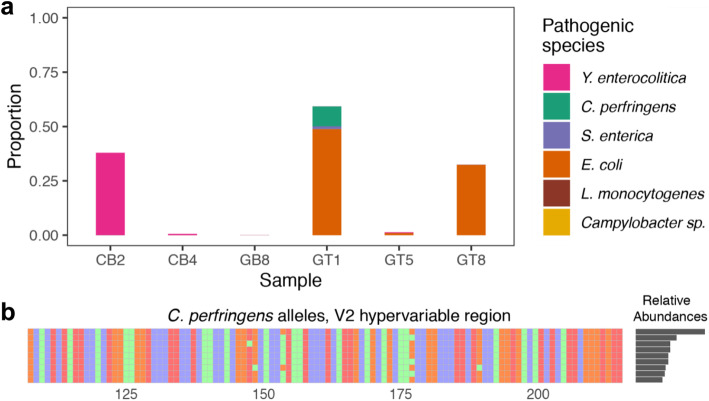
Results: LoopSeq reads are sufficiently long and accurate to identify microbial genes and species directly from complex samples. LoopSeq perfectly recovered the full diversity of 16S rRNA genes from known strains in a synthetic microbial community. Full-length LoopSeq reads had a per-base error rate of 0.005%, which exceeds the accuracy reported for other long-read sequencing technologies. 18S-ITS and genomic sequencing of fungal and bacterial isolates confirmed that LoopSeq sequencing maintains that accuracy for reads up to 6 kb in length. LoopSeq full-length 16S rRNA reads could accurately classify organisms down to the species level in rinsate from retail meat samples, and could differentiate strains within species identified by the CDC as potential foodborne pathogens.
Conclusions: The order-of-magnitude improvement in length and accuracy over standard Illumina amplicon sequencing achieved with LoopSeq enables accurate species-level and strain identification from complex- to low-biomass microbiome samples. The ability to generate accurate and long microbiome sequencing reads using standard short read sequencers will accelerate the building of quality microbial sequence databases and removes a significant hurdle on the path to precision microbial genomics. Video abstract.
Keywords: Amplicon sequencing; Long-read sequencing; Metagenomics; Synthetic long reads.
Conflict of interest statement
Michael Balamotis and Tuval Ben Yehezkel are employees of Loop Genomics, the vendor for the synthetic long-read sequencing technology analyzed in this manuscript.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
