

Polycomb repressive complex 2 in the driver's seat of childhood and young adult brain tumours
- PMID: 34092471
- PMCID: PMC8448921
- DOI: 10.1016/j.tcb.2021.05.006
Polycomb repressive complex 2 in the driver's seat of childhood and young adult brain tumours
Abstract
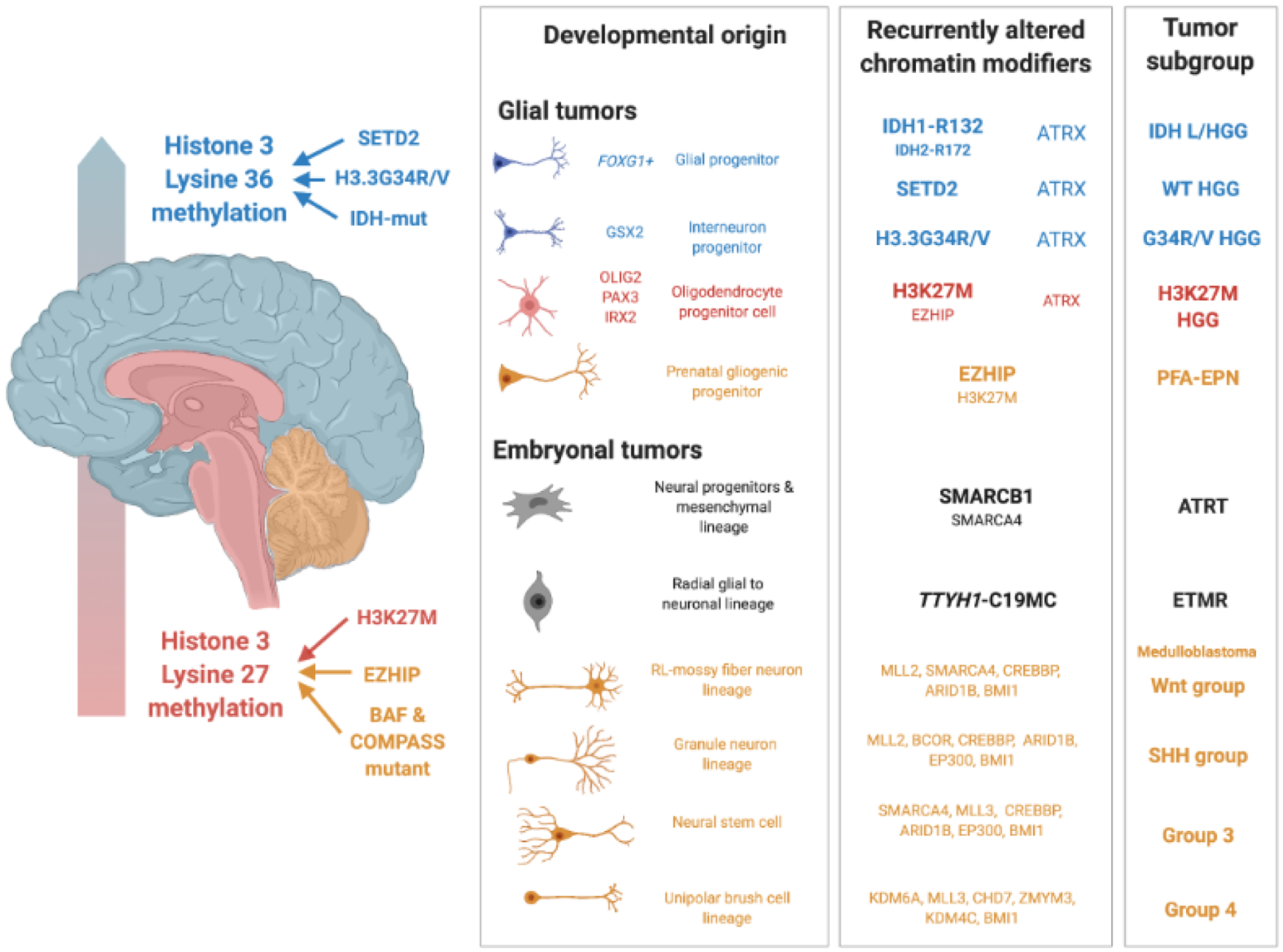
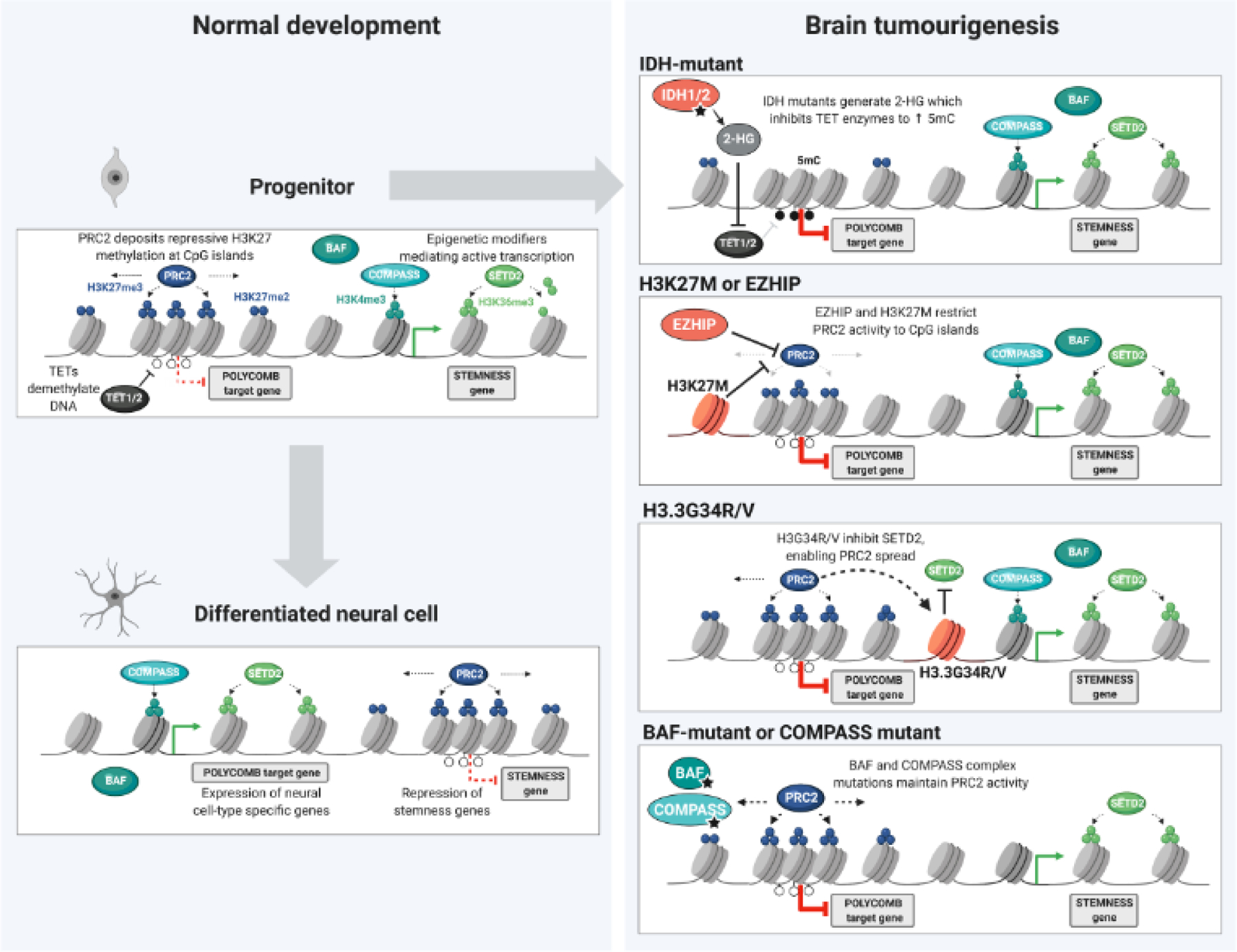
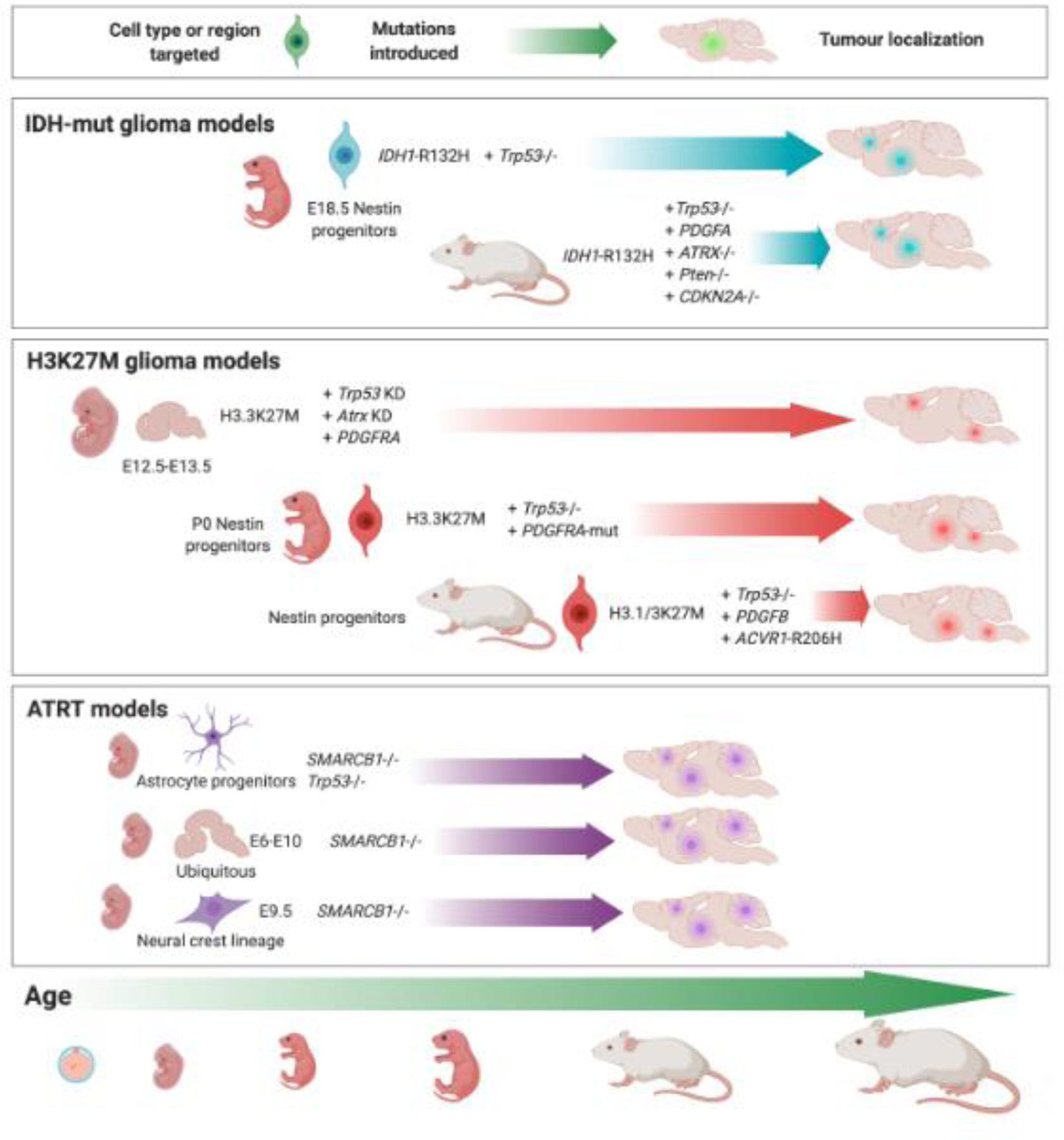
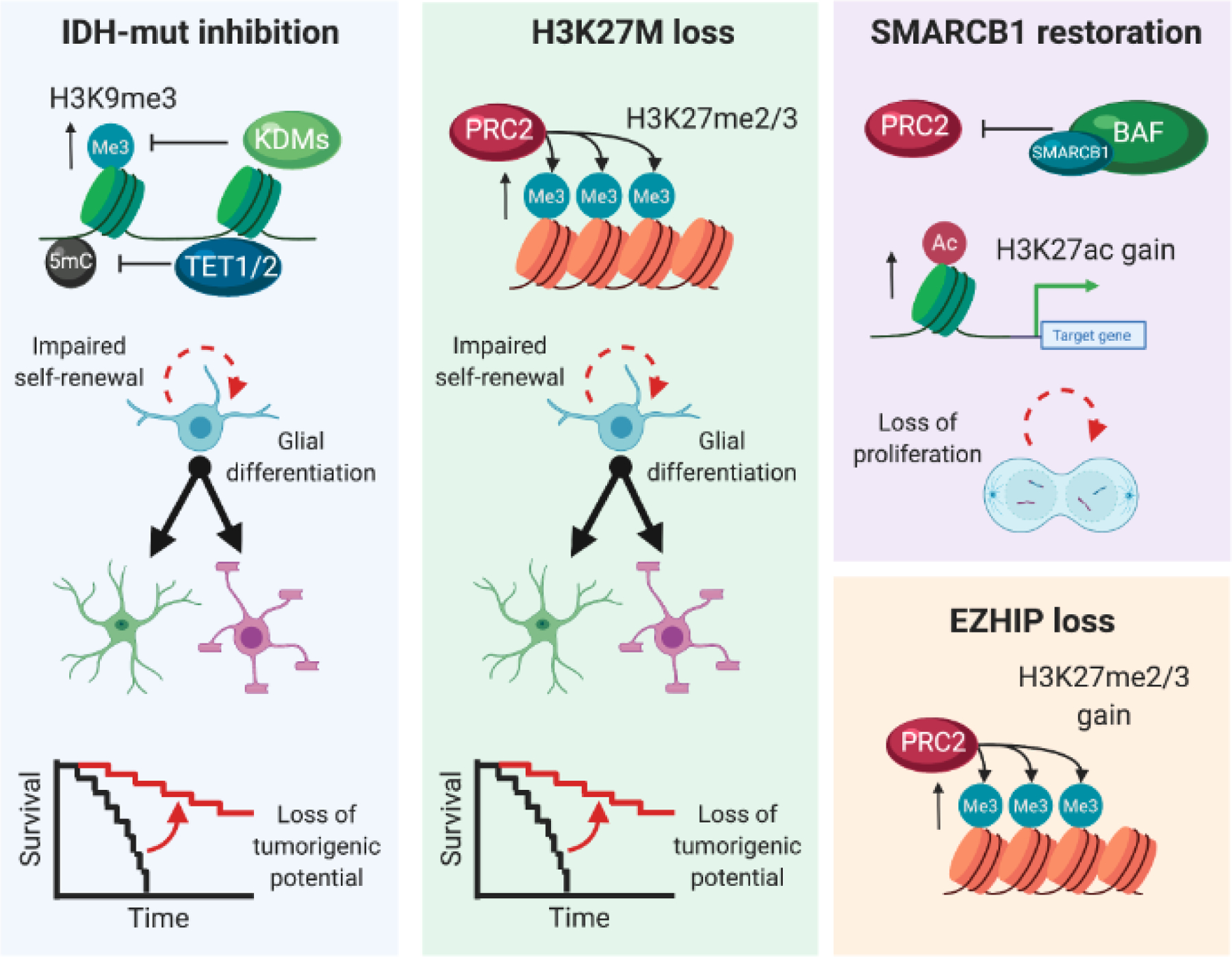
Deregulation of the epigenome underlies oncogenesis in numerous primary brain tumours in children and young adults. In this review, we describe how recurrent mutations in isocitrate dehydrogenases or histone 3 variants (oncohistones) in gliomas, expression of the oncohistone mimic enhancer of Zeste homologs inhibiting protein (EZHIP) in a subgroup of ependymoma, and epigenetic alterations in other embryonal tumours promote oncogenicity. We review the proposed mechanisms of cellular transformation, current tumorigenesis models and their link to development. We further stress the narrow developmental windows permissive to their oncogenic potential and how this may stem from converging effects deregulating polycomb repressive complex (PRC)2 function and targets. As altered chromatin states may be reversible, improved understanding of aberrant cancer epigenomes could orient the design of effective therapies.
Keywords: EZH inhibiting protein; epigenome; glioma; isocitrate dehydrogenases; polycomb repressive complex 2.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Louis DN et al. (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131 (6), 803–20. - PubMed
-
- Kleinman CL et al. (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46 (1), 39–44. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
