

Case Report: Identification of a de novo Microdeletion 1q44 in a Patient With Seizures and Developmental Delay
- PMID: 34093647
- PMCID: PMC8173053
- DOI: 10.3389/fgene.2021.648351
Case Report: Identification of a de novo Microdeletion 1q44 in a Patient With Seizures and Developmental Delay
Abstract
Objective: 1q44 microdeletion syndrome is difficult to diagnose due to the wide phenotypic spectrum and strong genetic heterogeneity. We explore the correlation between the chromosome microdeletions and phenotype in a child with 1q44 microdeletion syndrome, we collected the clinical features of the patient and combined them with adjacent copy number variation (CNV) regions previously reported. Methods: We collected the full medical history of the patient and summarized her clinical symptoms. Whole-exome sequencing (WES) and CapCNV analysis were performed with DNA extracted from both the patient's and her parents' peripheral blood samples. Fluorescent quantitative PCR (q-PCR) was performed for the use of verification to the CNV regions. Results: A 28.7 KB microdeletion was detected in the 1q44 region by whole-exome sequencing and low-depth whole-genome sequencing. The deleted region included the genes COX20 and HNRNPU. As verification, karyotype analysis showed no abnormality, and the results of qPCR were consistent with that of whole-exome sequencing and CapCNV analysis. Conclusion: The patient was diagnosed with 1q44 microdeletion syndrome with clinical and genetic analysis. Analyzing both whole-exome sequencing and CapCNV analysis can not only improve the diagnostic rate of clinically suspected syndromes that present with intellectual disability (ID) and multiple malformations but also support further study of the correlation between CNVs and clinical phenotypes. This study lays the foundation for the further study of the pathogenesis of complex diseases.
Keywords: 1q44 microdeletion syndrome; copy number variation; developmental delay; seizure; whole exon sequencing.
Copyright © 2021 Tung, Lu, Lin, Huang, Kim, Hu, Zhang and Zheng.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
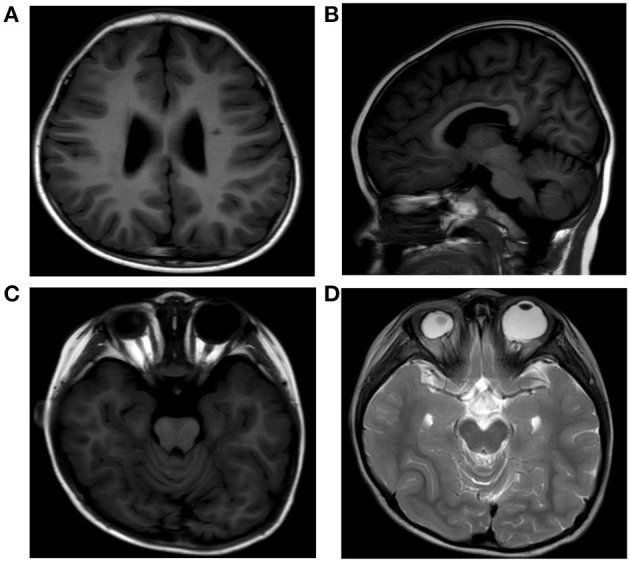
Figures




References
-
- Balak C., Belnap N., Ramsey K., Joss S., Devriendt K., Naymik M., et al. . (2018). A novel FBXO28 frameshift mutation in a child with developmental delay, dysmorphic features, and intractable epilepsy: a second gene that may contribute to the 1q41-q42 deletion phenotype. Am. J. Med. Gene. 176, 1549–1558. 10.1002/ajmg.a.38712 - DOI - PubMed
-
- Ballif B. C., Rosenfeld J. A., Traylor R., Theisen A., Bader P. I., Ladda R. L., et al. . (2012). High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum. Genet. 131, 145–156. 10.1007/s00439-011-1073-y - DOI - PubMed
-
- Boland E., Clayton-Smith J., Woo V. G., McKee S., Manson F. D., Medne L., et al. . (2007). Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 81, 292–303. 10.1086/519999 - DOI - PMC - PubMed
-
- Caliebe A., Kroes H. Y., van der Smagt J. J., Martin-Subero J. I., Tönnies H., van't, Slot R., et al. . (2010). Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. Eur. J. Med. Genet. 53, 179–185. 10.1016/j.ejmg.2010.04.001 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials