

HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes
- PMID: 34095222
- PMCID: PMC8170028
- DOI: 10.3389/fmolb.2021.663121
HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes
Abstract
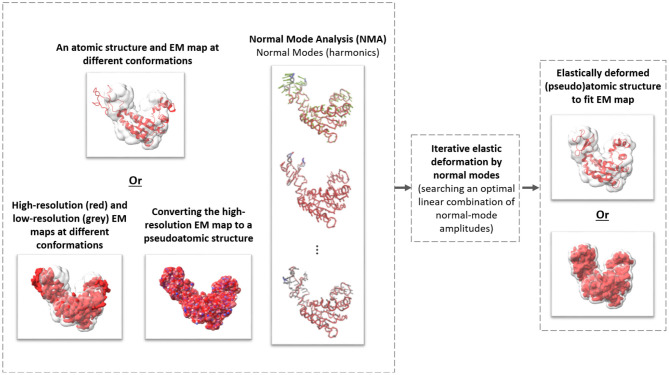
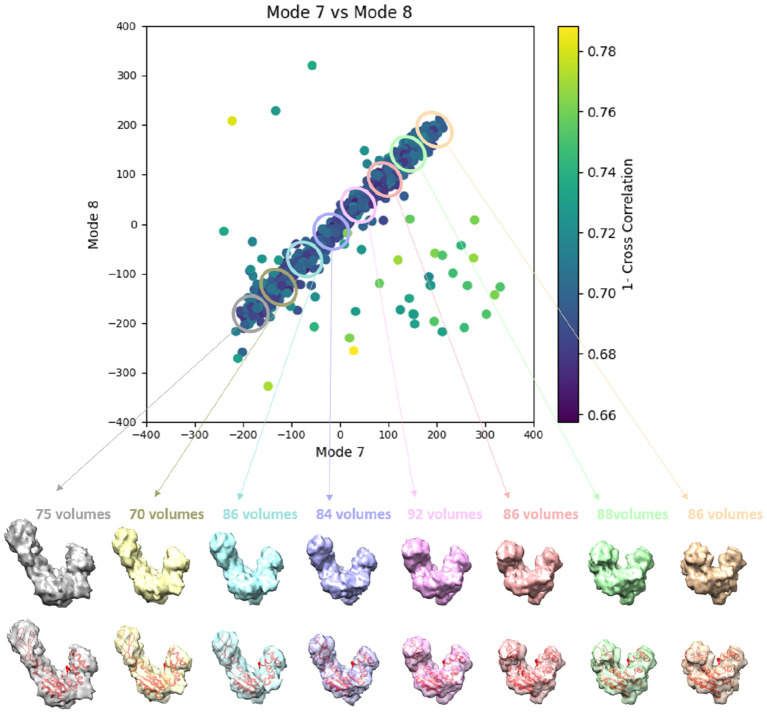
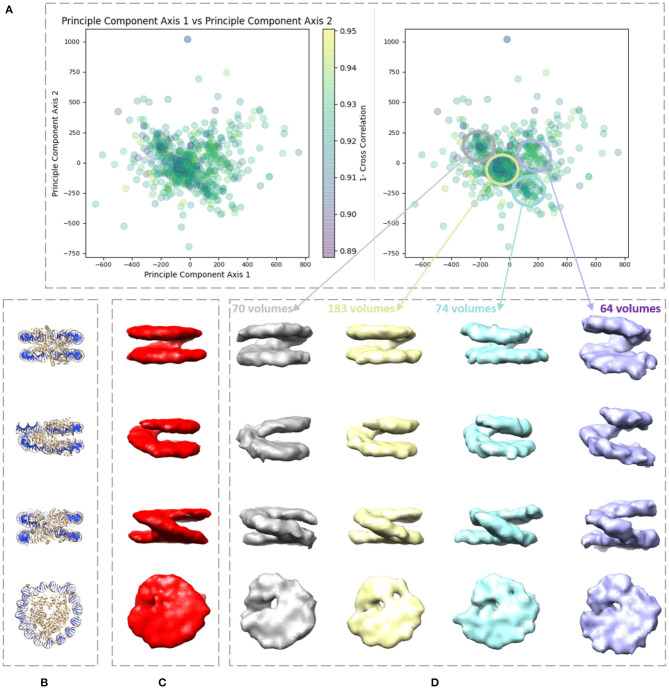
Cryogenic electron tomography (cryo-ET) allows structural determination of biomolecules in their native environment (in situ). Its potential of providing information on the dynamics of macromolecular complexes in cells is still largely unexploited, due to the challenges of the data analysis. The crowded cell environment and continuous conformational changes of complexes make difficult disentangling the data heterogeneity. We present HEMNMA-3D, which is, to the best of our knowledge, the first method for analyzing cryo electron subtomograms in terms of continuous conformational changes of complexes. HEMNMA-3D uses a combination of elastic and rigid-body 3D-to-3D iterative alignments of a flexible 3D reference (atomic structure or electron microscopy density map) to match the conformation, orientation, and position of the complex in each subtomogram. The elastic matching combines molecular mechanics simulation (Normal Mode Analysis of the 3D reference) and experimental, subtomogram data analysis. The rigid-body alignment includes compensation for the missing wedge, due to the limited tilt angle of cryo-ET. The conformational parameters (amplitudes of normal modes) of the complexes in subtomograms obtained through the alignment are processed to visualize the distribution of conformations in a space of lower dimension (typically, 2D or 3D) referred to as space of conformations. This allows a visually interpretable insight into the dynamics of the complexes, by calculating 3D averages of subtomograms with similar conformations from selected (densest) regions and by recording movies of the 3D reference's displacement along selected trajectories through the densest regions. We describe HEMNMA-3D and show its validation using synthetic datasets. We apply HEMNMA-3D to an experimental dataset describing in situ nucleosome conformational variability. HEMNMA-3D software is available freely (open-source) as part of ContinuousFlex plugin of Scipion V3.0 (http://scipion.i2pc.es).
Keywords: continuous conformational changes; cryo electron tomography; flexible-reference alignment; normal mode analysis; nucleosomes in situ.
Copyright © 2021 Harastani, Eltsov, Leforestier and Jonic.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures








Similar articles
-
Hybrid Electron Microscopy Normal Mode Analysis with Scipion.Protein Sci. 2020 Jan;29(1):223-236. doi: 10.1002/pro.3772. Epub 2019 Nov 20. Protein Sci. 2020. PMID: 31693263 Free PMC article.
-
ContinuousFlex: Software package for analyzing continuous conformational variability of macromolecules in cryo electron microscopy and tomography data.J Struct Biol. 2022 Dec;214(4):107906. doi: 10.1016/j.jsb.2022.107906. Epub 2022 Oct 13. J Struct Biol. 2022. PMID: 36244611
-
TomoFlow: Analysis of Continuous Conformational Variability of Macromolecules in Cryogenic Subtomograms based on 3D Dense Optical Flow.J Mol Biol. 2022 Jan 30;434(2):167381. doi: 10.1016/j.jmb.2021.167381. Epub 2021 Nov 27. J Mol Biol. 2022. PMID: 34848215
-
Cryo-electron tomography for the structural study of mitochondrial translation.Tissue Cell. 2019 Apr;57:129-138. doi: 10.1016/j.tice.2018.08.009. Epub 2018 Aug 29. Tissue Cell. 2019. PMID: 30197222 Review.
-
Cryo-Electron Tomography and Subtomogram Averaging.Methods Enzymol. 2016;579:329-67. doi: 10.1016/bs.mie.2016.04.014. Epub 2016 Jun 22. Methods Enzymol. 2016. PMID: 27572733 Review.
Cited by
-
A method for restoring signals and revealing individual macromolecule states in cryo-ET, REST.Nat Commun. 2023 May 22;14(1):2937. doi: 10.1038/s41467-023-38539-w. Nat Commun. 2023. PMID: 37217501 Free PMC article.
-
CryoDRGN-AI: Neural ab initio reconstruction of challenging cryo-EM and cryo-ET datasets.bioRxiv [Preprint]. 2025 Apr 28:2024.05.30.596729. doi: 10.1101/2024.05.30.596729. bioRxiv. 2025. Update in: Nat Methods. 2025 Jul;22(7):1486-1494. doi: 10.1038/s41592-025-02720-4. PMID: 38854058 Free PMC article. Updated. Preprint.
-
Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing.Int J Mol Sci. 2023 Sep 18;24(18):14245. doi: 10.3390/ijms241814245. Int J Mol Sci. 2023. PMID: 37762547 Free PMC article.
-
Real-space heterogeneous reconstruction, refinement, and disentanglement of CryoEM conformational states with HetSIREN.Nat Commun. 2025 Apr 22;16(1):3751. doi: 10.1038/s41467-025-59135-0. Nat Commun. 2025. PMID: 40263313 Free PMC article.
-
CryoDRGN-ET: deep reconstructing generative networks for visualizing dynamic biomolecules inside cells.Nat Methods. 2024 Aug;21(8):1537-1545. doi: 10.1038/s41592-024-02340-4. Epub 2024 Jul 18. Nat Methods. 2024. PMID: 39025970
References
-
- Berghen F. V., Bersini H. (2005). Condor, a new parallel, constrained extension of Powell's Uobyqa algorithm: experimental results and comparison with the dfo algorithm. J. Comput. Appl. Math. 181, 157–175. 10.1016/j.cam.2004.11.029 - DOI
LinkOut - more resources
Full Text Sources