

Innovative Treatments for Rare Anemias
- PMID: 34095760
- PMCID: PMC8171369
- DOI: 10.1097/HS9.0000000000000576
Innovative Treatments for Rare Anemias
Abstract
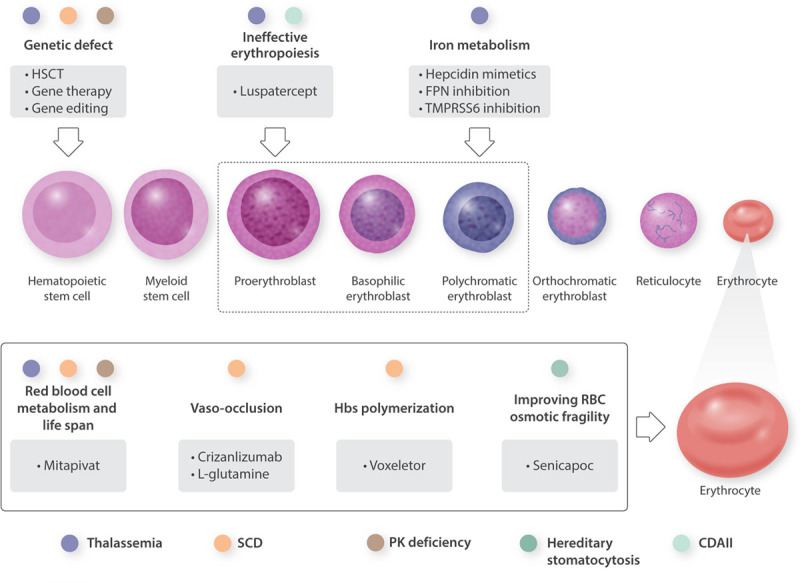
Rare anemias (RA) are mostly hereditary disorders with low prevalence and a broad spectrum of clinical severity, affecting different stages of erythropoiesis or red blood cell components. RA often remains underdiagnosed or misdiagnosed, and treatment options have been limited to supportive care for many years. During the last decades, the elucidation of the molecular mechanisms underlying several RA paved the way for developing new treatments. Innovative treatments other than supportive care and allogeneic bone marrow transplantation are currently in clinical trials for β-thalassemias, sickle cell disease (SCD), and congenital hemolytic anemias. Recently, luspatercept, an activin receptor ligand trap targeting ineffective erythropoiesis, has been approved as the first pharmacological treatment for transfusion-dependent β-thalassemia. L-glutamine, voxelotor, and crizanlizumab are new drugs approved SCD, targeting different steps of the complex pathophysiological mechanism. Gene therapy represents an innovative and encouraging strategy currently under evaluation in several RA and recently approved for β-thalassemia. Moreover, the advent of gene-editing technologies represents an additional option, mainly focused on correcting the defective gene or editing the expression of genes that regulate fetal hemoglobin synthesis. In this review, we aim to update the status of innovative treatments and the ongoing trials and discuss RA treatments' future directions. Interestingly, several molecules that showed promising results for treating one of these disorders are now under evaluation in the others. In the near future, the management of RA will probably consist of polypharmacotherapy tailored to patients' characteristics.
Copyright © 2021 the Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the European Hematology Association.
Figures
References
-
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1545–1602. - PMC - PubMed
-
- Brissot P, Bernard DG, Brissot E, et al. . Rare anemias due to genetic iron metabolism defects. Mutat Res. 2018; 777:52–63. - PubMed
-
- Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. eds. Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). 3rd ed. Nicosia, Cyprus: Thalassaemia International Federation; TIF Publication No. 20; 2014:1–253. - PubMed
-
- Taher A, Musallam KM, Cappellini MD. eds. Guidelines for the Management of Non-Transfusion Dependent Thalassaemia (NTDT). 3nd ed. Nicosia, Cyprus: Thalassaemia International Federation; TIF Publication No. 22; 2017:1–117. - PubMed