

Research in Sickle Cell Disease: From Bedside to Bench to Bedside
- PMID: 34095767
- PMCID: PMC8171370
- DOI: 10.1097/HS9.0000000000000584
Research in Sickle Cell Disease: From Bedside to Bench to Bedside
Abstract
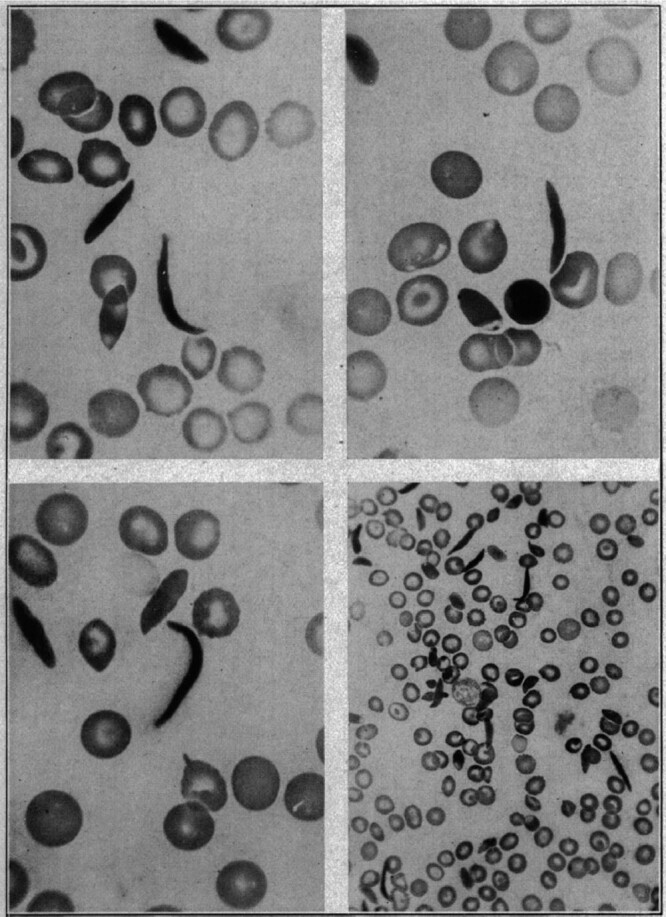
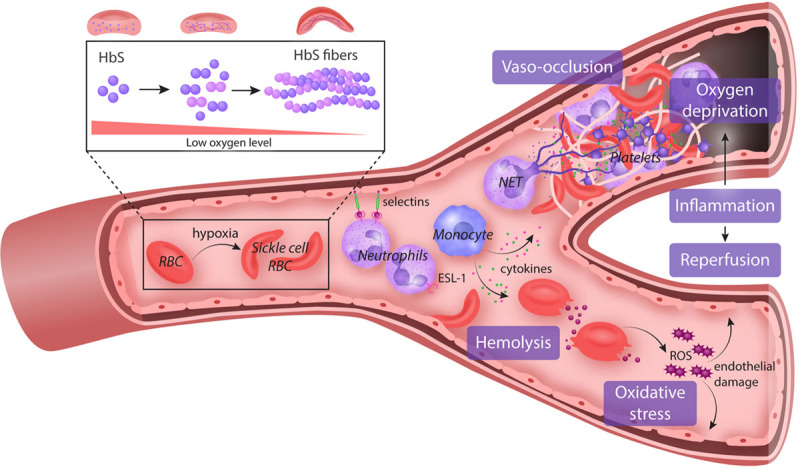
Sickle cell disease (SCD) is an exemplar of bidirectional translational research, starting with a remarkable astute observation of the abnormally shaped red blood cells that motivated decades of bench research that have now translated into new drugs and genetic therapies. Introduction of hydroxyurea (HU) therapy, the only SCD-modifying treatment for >30 years and now standard care, was initiated through another clinical observation by a pediatrician. While the clinical efficacy of HU is primarily due to its fetal hemoglobin (HbF) induction, the exact mechanism of how it increases HbF remains not fully understood. Unraveling of the molecular mechanism of how HU increases HbF has provided insights on the development of new HbF-reactivating agents in the pipeline. HU has other salutary effects, reduction of cellular adhesion to the vascular endothelium and inflammation, and dissecting these mechanisms has informed bench-both cellular and animal-research for development of the 3 recently approved agents: endari, voxelotor, and crizanlizumab; truly, a bidirectional bench to bedside translation. Decades of research to understand the mechanisms of fetal to adult hemoglobin have also culminated in promising anti-sickling genetic therapies and the first-in-human studies of reactivating an endogenous (γ-globin) gene HBG utilizing innovative genomic approaches.
Copyright © 2021 the Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the European Hematology Association.
Figures
References
-
- Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. JAMA. 2014; 312:1063. - PubMed
-
- Pauling L, Itano HA. Sickle cell anemia a molecular disease. Science. 1949; 110:543–548. - PubMed
-
- Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956; 178:792–794. - PubMed
-
- Goldstein J, Konigsberg W, Hill RJ. The structure of human hemoglobin. VI. The sequence of amino acids in the tryptic peptides of the beta chain. J Biol Chem. 1963; 238:2016–2027. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources