

Anderson-Fabry Disease: From Endothelial Dysfunction to Emerging Therapies
- PMID: 34095851
- PMCID: PMC8137293
- DOI: 10.1155/2021/5548445
Anderson-Fabry Disease: From Endothelial Dysfunction to Emerging Therapies
Abstract
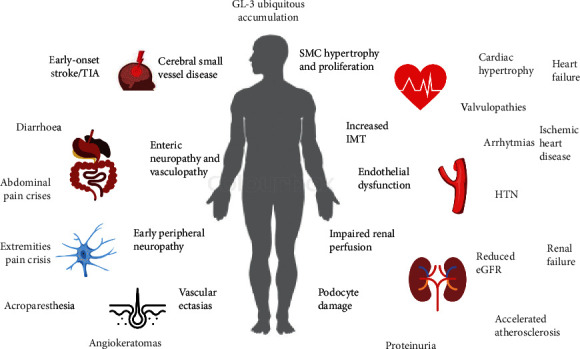
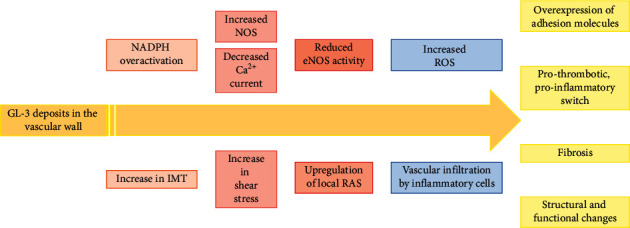
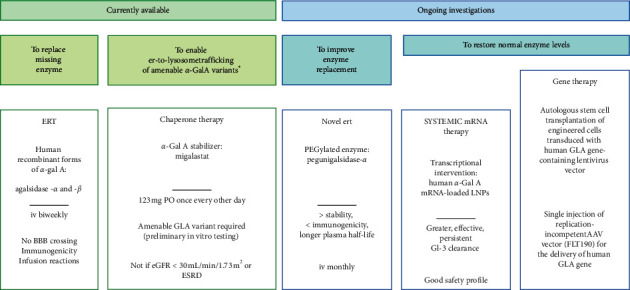
The Anderson-Fabry disease is a rare, X-linked, multisystemic, progressive lysosomal storage disease caused by α-galactosidase A total or partial deficiency. The resulting syndrome is mainly characterized by early-onset autonomic neuropathy and life-threatening multiorgan involvement, including renal insufficiency, heart disease, and early stroke. The enzyme deficiency leads to tissue accumulation of the glycosphingolipid globotriaosylceramide and its analogues, but the mechanisms linking such accumulation to organ damage are only partially understood. In contrast, enzyme replacement and chaperone therapies are already fully available to patients and allow substantial amelioration of quality and quantity of life. Substrate reduction, messenger ribonucleic acid (mRNA)-based, and gene therapies are also on the horizon. In this review, the clinical scenario and molecular aspects of Anderson-Fabry disease are described, along with updates on disease mechanisms and emerging therapies.
Copyright © 2021 Cosimo A. Stamerra et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
-
- Fabry J. Ein beitrag zur kenntniss der purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica hebrae) Archiv für Dermatologie und Syphilis. 1898;43(1):187–200. doi: 10.1007/bf01986897. - DOI
-
- Anderson W. A case of “angeio-keratoma”. British Journal of Dermatology. 1898;10(4):113–117. doi: 10.1111/j.1365-2133.1898.tb16317.x. - DOI
Publication types
LinkOut - more resources
Full Text Sources