

Addressing opioid tolerance and opioid-induced hypersensitivity: Recent developments and future therapeutic strategies
- PMID: 34096178
- PMCID: PMC8181203
- DOI: 10.1002/prp2.789
Addressing opioid tolerance and opioid-induced hypersensitivity: Recent developments and future therapeutic strategies
Abstract
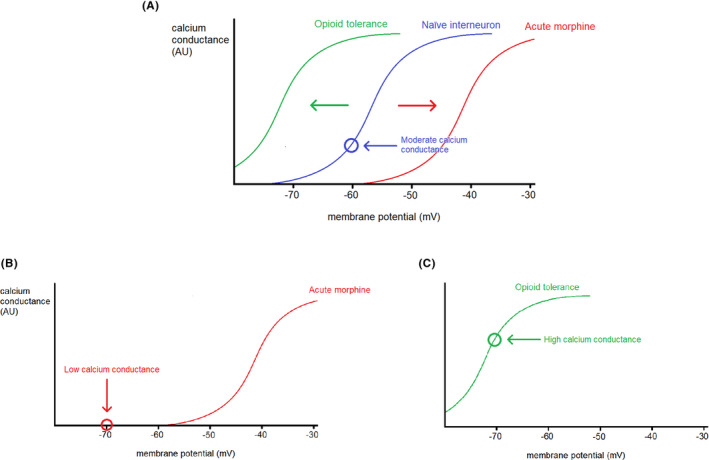
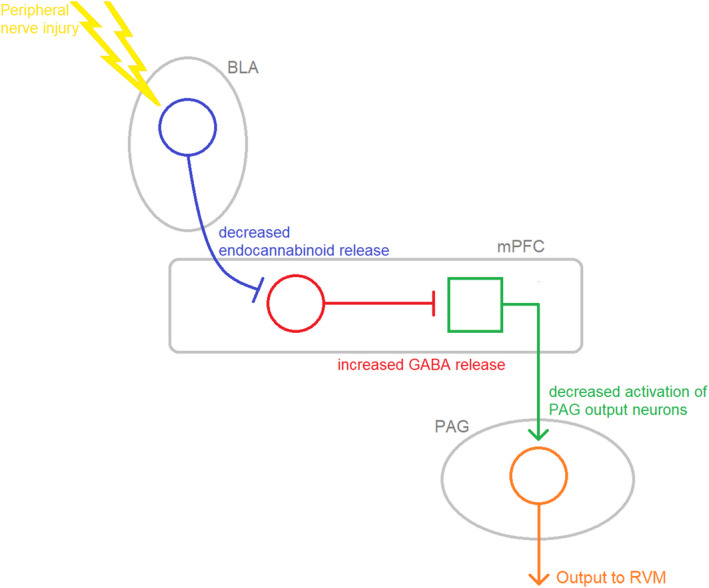
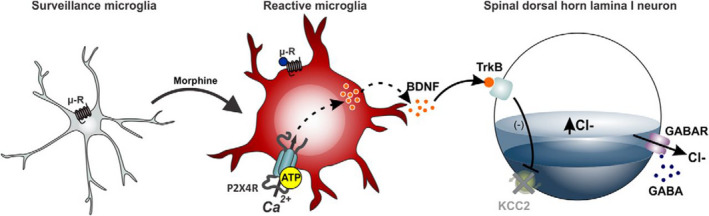
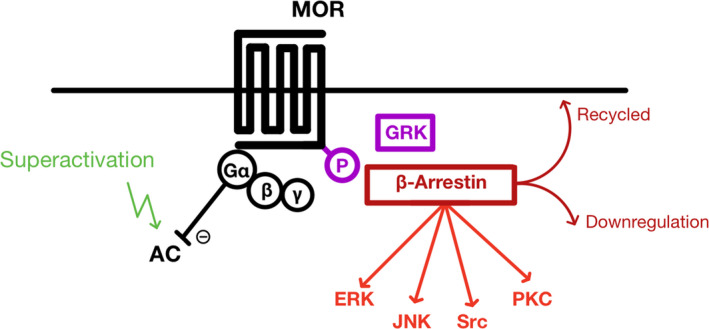
Opioids are a commonly prescribed and efficacious medication for the treatment of chronic pain but major side effects such as addiction, respiratory depression, analgesic tolerance, and paradoxical pain hypersensitivity make them inadequate and unsafe for patients requiring long-term pain management. This review summarizes recent advances in our understanding of the outcomes of chronic opioid administration to lay the foundation for the development of novel pharmacological strategies that attenuate opioid tolerance and hypersensitivity; the two main physiological mechanisms underlying the inadequacies of current therapeutic strategies. We also explore mechanistic similarities between the development of neuropathic pain states, opioid tolerance, and hypersensitivity which may explain opioids' lack of efficacy in certain patients. The findings challenge the current direction of analgesic research in developing non-opioid alternatives and we suggest that improving opioids, rather than replacing them, will be a fruitful avenue for future research.
Keywords: analgesia; opioid receptors; opioid tolerance; opioid-induced hypersensitivity; opioids; pain.
© 2021 The Authors. Pharmacology Research & Perspectives published by British Pharmacological Society and American Society for Pharmacology and Experimental Therapeutics and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Opioid Receptors.Annu Rev Med. 2016;67:433-51. doi: 10.1146/annurev-med-062613-093100. Epub 2015 Aug 26. Annu Rev Med. 2016. PMID: 26332001 Review.
-
[Development of opioid tolerance -- molecular mechanisms and clinical consequences].Anasthesiol Intensivmed Notfallmed Schmerzther. 2003 Jan;38(1):14-26. doi: 10.1055/s-2003-36558. Anasthesiol Intensivmed Notfallmed Schmerzther. 2003. PMID: 12522725 Review. German.
-
Opioid analgesia: recent developments.Curr Opin Support Palliat Care. 2020 Jun;14(2):112-117. doi: 10.1097/SPC.0000000000000495. Curr Opin Support Palliat Care. 2020. PMID: 32304400 Review.
-
Opioids and the management of chronic severe pain in the elderly: consensus statement of an International Expert Panel with focus on the six clinically most often used World Health Organization Step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone).Pain Pract. 2008 Jul-Aug;8(4):287-313. doi: 10.1111/j.1533-2500.2008.00204.x. Epub 2008 May 23. Pain Pract. 2008. PMID: 18503626
-
Review of the effect of opioid-related side effects on the undertreatment of moderate to severe chronic non-cancer pain: tapentadol, a step toward a solution?Curr Med Res Opin. 2010 Jul;26(7):1677-84. doi: 10.1185/03007995.2010.483941. Curr Med Res Opin. 2010. PMID: 20465361 Review.
Cited by
-
The Burden of Metastatic Cancer-Induced Bone Pain: A Narrative Review.J Pain Res. 2022 Oct 25;15:3399-3412. doi: 10.2147/JPR.S371337. eCollection 2022. J Pain Res. 2022. PMID: 36317162 Free PMC article. Review.
-
Co-Administration of nalbuphine to improve morphine tolerance in mice with bone cancer pain.Mol Pain. 2023 Jan-Dec;19:17448069231178741. doi: 10.1177/17448069231178741. Mol Pain. 2023. PMID: 37226458 Free PMC article.
-
State and Future Science of Opioids and Potential of Biased-ligand Technology in the Management of Acute Pain After Burn Injury.J Burn Care Res. 2023 May 2;44(3):524-534. doi: 10.1093/jbcr/irad004. J Burn Care Res. 2023. PMID: 36638083 Free PMC article. Review.
-
Underutilized treatments for patients with refractory cancer pain: a qualitative study assessing the use of intrathecal drug delivery devices in the United Kingdom compared to alternative treatments in cancer pain management.Front Pain Res (Lausanne). 2025 Feb 20;6:1481245. doi: 10.3389/fpain.2025.1481245. eCollection 2025. Front Pain Res (Lausanne). 2025. PMID: 40051773 Free PMC article.
-
Machine Learning to Predict Successful Opioid Dose Reduction or Stabilization After Spinal Cord Stimulation.Neurosurgery. 2022 Aug 1;91(2):272-279. doi: 10.1227/neu.0000000000001969. Epub 2022 Apr 8. Neurosurgery. 2022. PMID: 35384918 Free PMC article.
References
-
- IASP Task Force on Taxonomy . Classification of Chronic Pain. 2nd ed. (Revised). In Merskey H, Bogduk N eds. Washington, DC: IASP Press; 1994. https://www.iasp‐pain.org/Education/Content.aspx?ItemNumber=1698. Accessed April 7, 2020.
-
- Kirsch B, Berdine H, Zablotsky D, Wenzel G, Meyer C. Management strategy: identifying pain as the fifth vital sign University. Vet Health Syst J. 2000;49‐59.
-
- Lee M, Silverman SM, Hansen H, Patel VB, Manchikanti L. A comprehensive review of opioid‐induced hyperalgesia. Pain Phys. 2011;14(2):145‐161. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
