

The HGR motif is the antiangiogenic determinant of vasoinhibin: implications for a therapeutic orally active oligopeptide
- PMID: 34097181
- PMCID: PMC8813873
- DOI: 10.1007/s10456-021-09800-x
The HGR motif is the antiangiogenic determinant of vasoinhibin: implications for a therapeutic orally active oligopeptide
Abstract
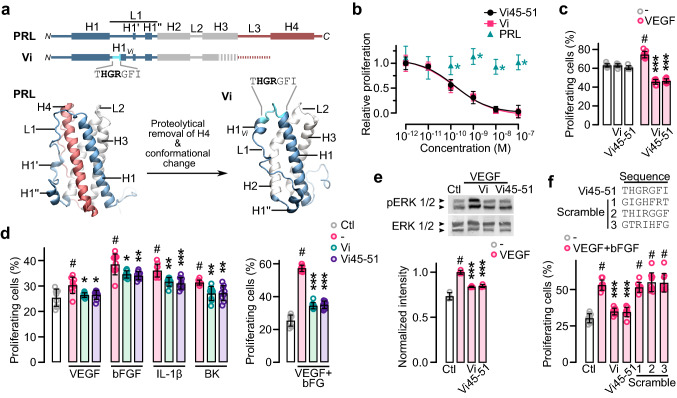
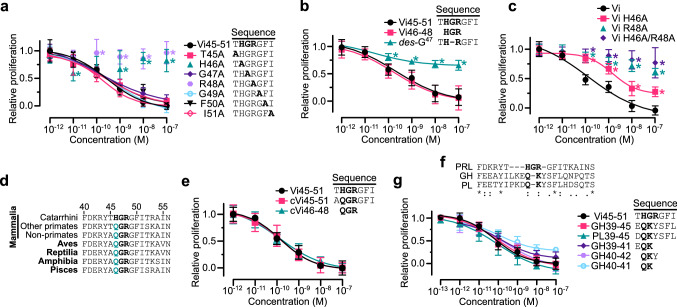
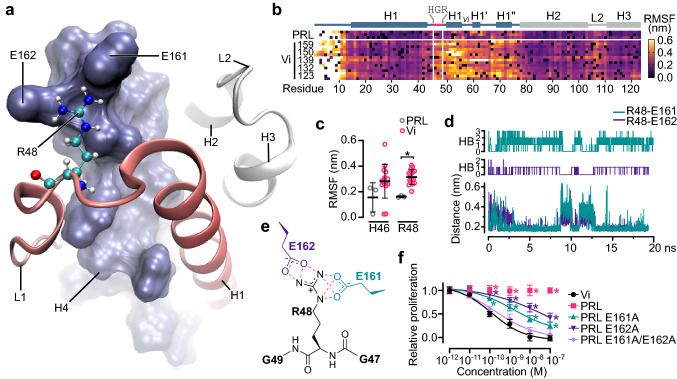
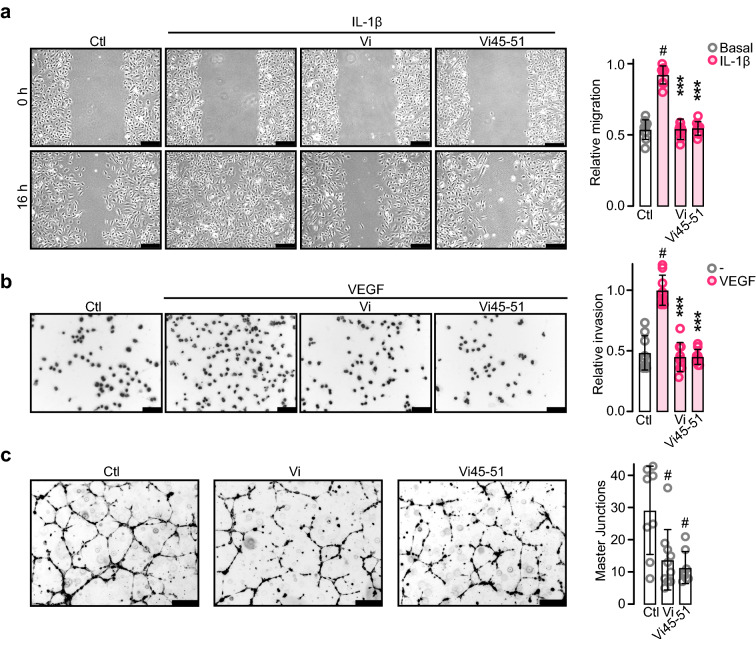
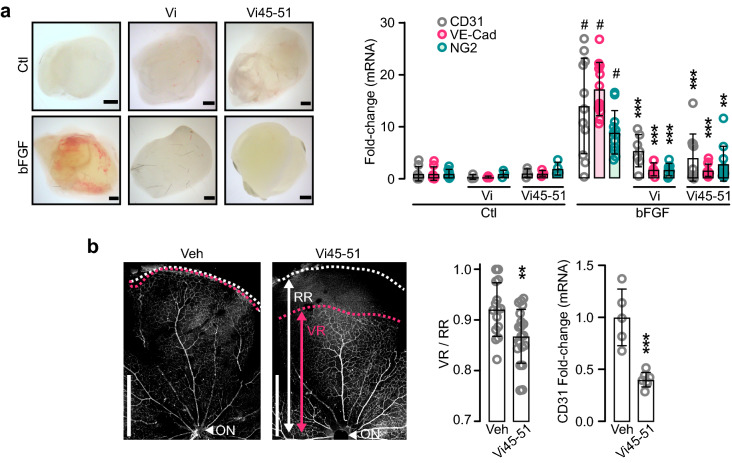
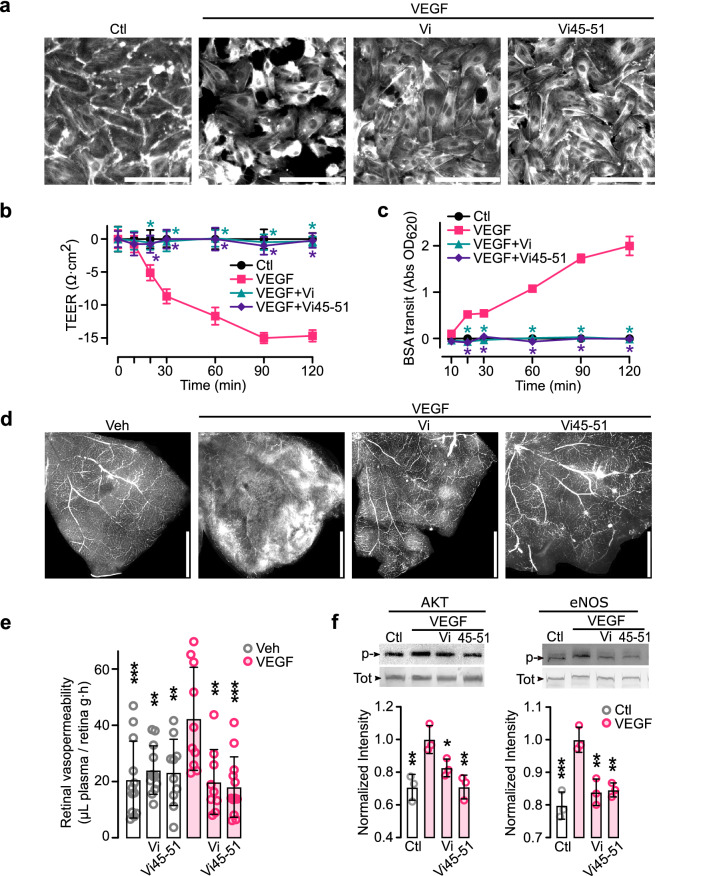
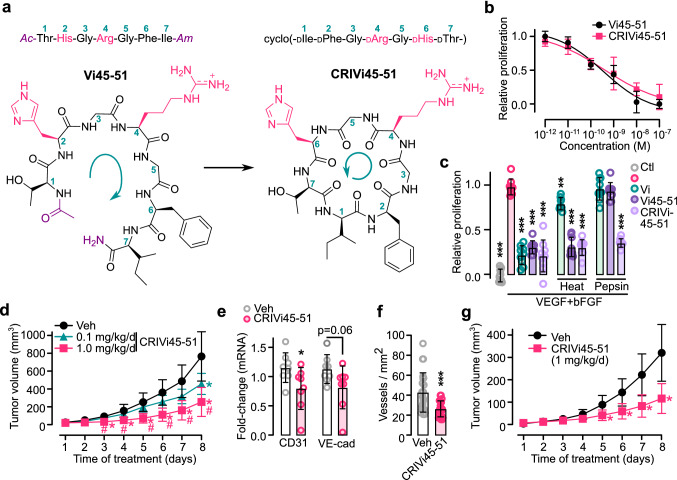
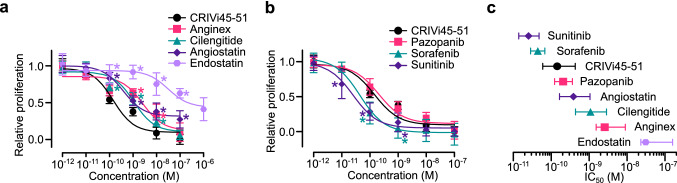
The hormone prolactin acquires antiangiogenic and antivasopermeability properties after undergoing proteolytic cleavage to vasoinhibin, an endogenous prolactin fragment of 123 or more amino acids that inhibits the action of multiple proangiogenic factors. Preclinical and clinical evidence supports the therapeutic potential of vasoinhibin against angiogenesis-related diseases including diabetic retinopathy, peripartum cardiomyopathy, rheumatoid arthritis, and cancer. However, the use of vasoinhibin in the clinic has been limited by difficulties in its production. Here, we removed this barrier to using vasoinhibin as a therapeutic agent by showing that a short linear motif of just three residues (His46-Gly47-Arg48) (HGR) is the functional determinant of vasoinhibin. The HGR motif is conserved throughout evolution, its mutation led to vasoinhibin loss of function, and oligopeptides containing this sequence inhibited angiogenesis and vasopermeability with the same potency as whole vasoinhibin. Furthermore, the oral administration of an optimized cyclic retro-inverse vasoinhibin heptapeptide containing HGR inhibited melanoma tumor growth and vascularization in mice and exhibited equal or higher antiangiogenic potency than other antiangiogenic molecules currently used as anti-cancer drugs in the clinic. Finally, by unveiling the mechanism that obscures the HGR motif in prolactin, we anticipate the development of vasoinhibin-specific antibodies to solve the on-going challenge of measuring endogenous vasoinhibin levels for diagnostic and interventional purposes, the design of vasoinhibin antagonists for managing insufficient angiogenesis, and the identification of putative therapeutic proteins containing HGR.
Keywords: 16K prolactin; Angiogenesis; Melanoma; Oligopeptide; Retina; Vasoinhibin; Vasopermeability.
© 2021. The Author(s).
Conflict of interest statement
The authors declare the following competing interests: C.C., J.P.R., M.Z., G.M.E., J.T., and T.B. are inventors of submitted Mexican (MX/E/2019/079075) and multinational (PCT/EP2020/069154) patent applications. The
Figures
Similar articles
-
Vasoinhibin's Apoptotic, Inflammatory, and Fibrinolytic Actions Are in a Motif Different From Its Antiangiogenic HGR Motif.Endocrinology. 2023 Dec 23;165(2):bqad185. doi: 10.1210/endocr/bqad185. Endocrinology. 2023. PMID: 38057149 Free PMC article.
-
Thrombin Cleaves Prolactin Into a Potent 5.6-kDa Vasoinhibin: Implication for Tissue Repair.Endocrinology. 2021 Dec 1;162(12):bqab177. doi: 10.1210/endocr/bqab177. Endocrinology. 2021. PMID: 34418052
-
Development of Vasoinhibin-Specific Monoclonal Antibodies.Front Endocrinol (Lausanne). 2021 Apr 20;12:645085. doi: 10.3389/fendo.2021.645085. eCollection 2021. Front Endocrinol (Lausanne). 2021. PMID: 33959096 Free PMC article.
-
Prolactin and vasoinhibin are endogenous players in diabetic retinopathy revisited.Front Endocrinol (Lausanne). 2022 Sep 9;13:994898. doi: 10.3389/fendo.2022.994898. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36157442 Free PMC article. Review.
-
From Bench to Bedside: Translating the Prolactin/Vasoinhibin Axis.Front Endocrinol (Lausanne). 2017 Dec 11;8:342. doi: 10.3389/fendo.2017.00342. eCollection 2017. Front Endocrinol (Lausanne). 2017. PMID: 29321761 Free PMC article. Review.
Cited by
-
Vasoinhibin is Generated by the Renin-angiotensin System.Endocrinology. 2025 Feb 5;166(3):bqaf023. doi: 10.1210/endocr/bqaf023. Endocrinology. 2025. PMID: 39903548 Free PMC article.
-
14K prolactin derived 14-mer antiangiogenic peptide targets bradykinin-/nitric oxide-cGMP-dependent angiogenesis.FEBS Open Bio. 2024 Dec;14(12):2072-2085. doi: 10.1002/2211-5463.13895. Epub 2024 Sep 23. FEBS Open Bio. 2024. PMID: 39312470 Free PMC article.
-
Vasoinhibin's Apoptotic, Inflammatory, and Fibrinolytic Actions Are in a Motif Different From Its Antiangiogenic HGR Motif.Endocrinology. 2023 Dec 23;165(2):bqad185. doi: 10.1210/endocr/bqad185. Endocrinology. 2023. PMID: 38057149 Free PMC article.
-
Human Placental Tissue Contains A Placental Lactogen-Derived Vasoinhibin.J Endocr Soc. 2022 Feb 20;6(4):bvac029. doi: 10.1210/jendso/bvac029. eCollection 2022 Apr 1. J Endocr Soc. 2022. PMID: 35265784 Free PMC article.
-
Immunometric and functional measurement of endogenous vasoinhibin in human sera.Front Endocrinol (Lausanne). 2024 Apr 29;15:1345996. doi: 10.3389/fendo.2024.1345996. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 38742198 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
