

Protein haploinsufficiency drivers identify MYBPC3 variants that cause hypertrophic cardiomyopathy
- PMID: 34097875
- PMCID: PMC8260873
- DOI: 10.1016/j.jbc.2021.100854
Protein haploinsufficiency drivers identify MYBPC3 variants that cause hypertrophic cardiomyopathy
Abstract
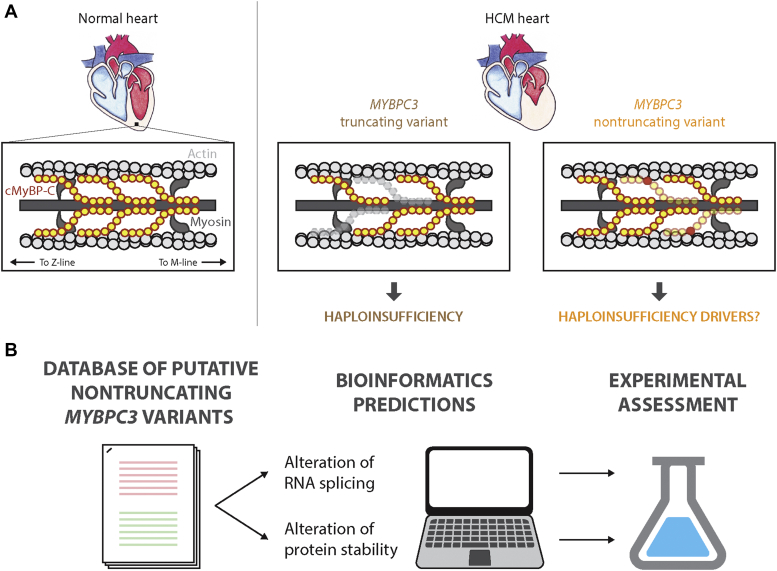
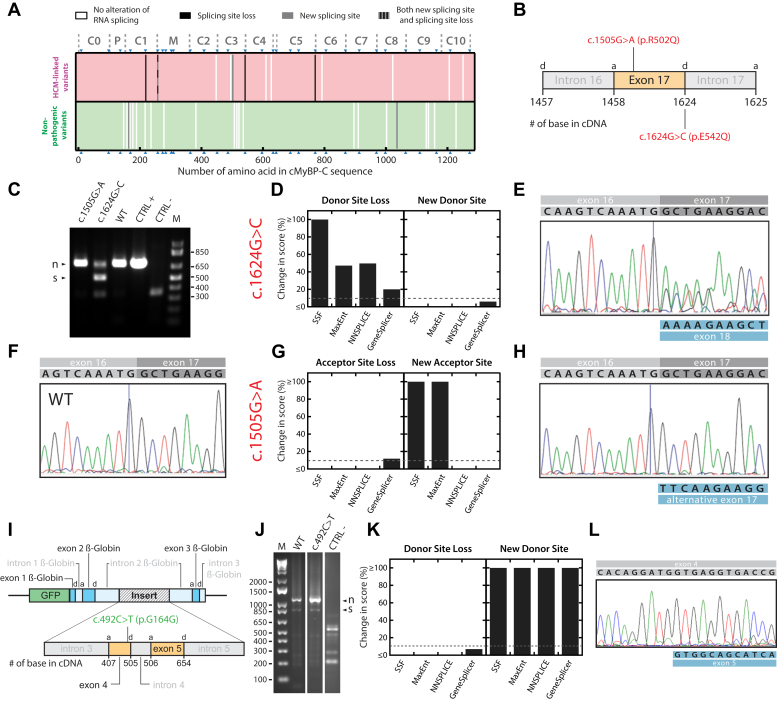
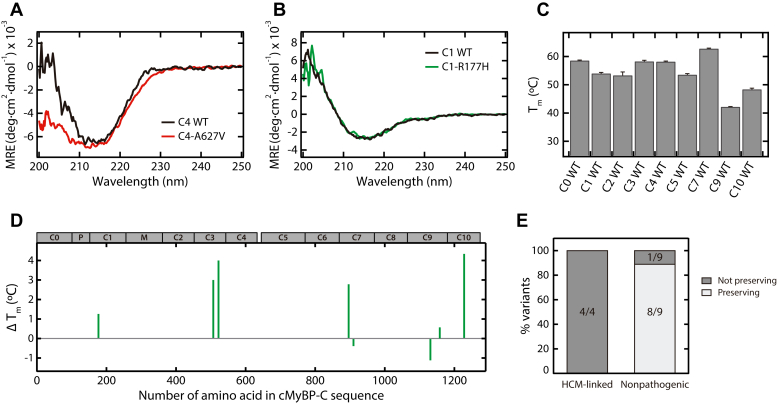
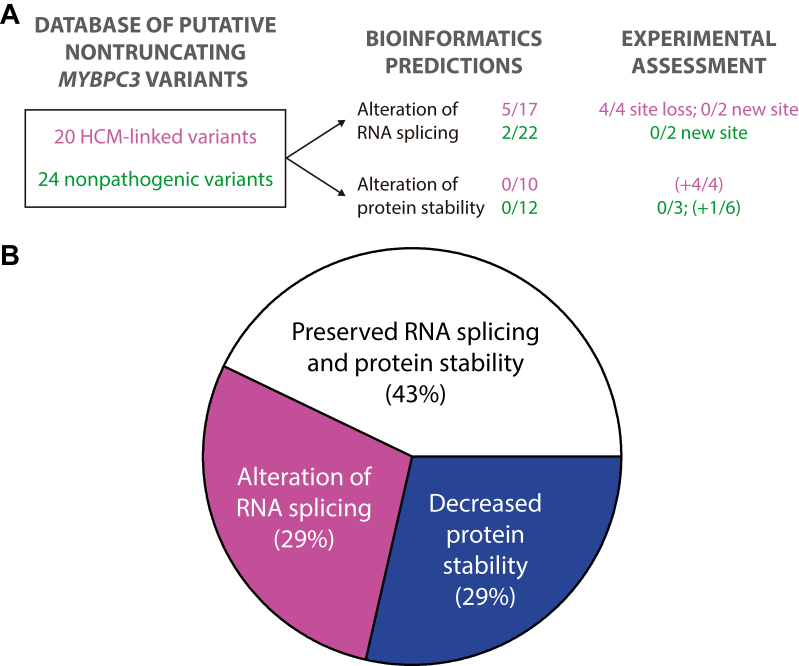
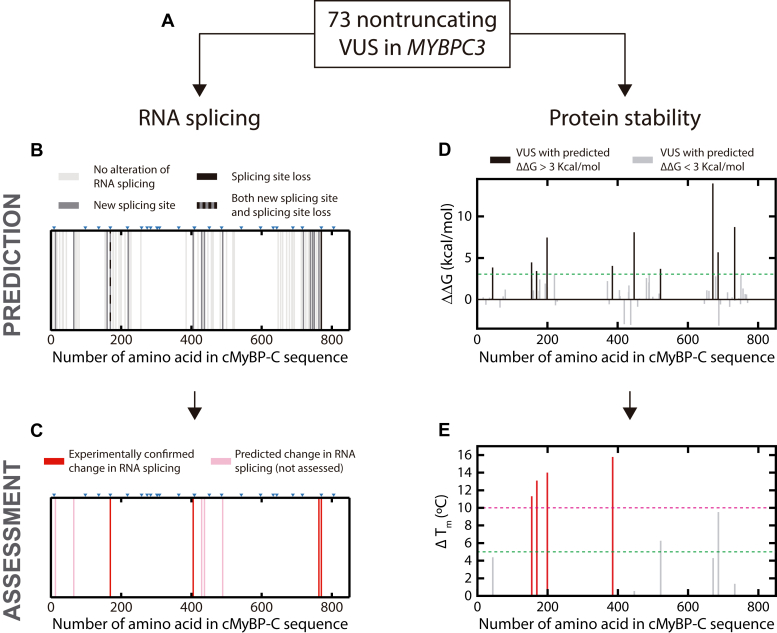
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease. Variants in MYBPC3, the gene encoding cardiac myosin-binding protein C (cMyBP-C), are the leading cause of HCM. However, the pathogenicity status of hundreds of MYBPC3 variants found in patients remains unknown, as a consequence of our incomplete understanding of the pathomechanisms triggered by HCM-causing variants. Here, we examined 44 nontruncating MYBPC3 variants that we classified as HCM-linked or nonpathogenic according to cosegregation and population genetics criteria. We found that around half of the HCM-linked variants showed alterations in RNA splicing or protein stability, both of which can lead to cMyBP-C haploinsufficiency. These protein haploinsufficiency drivers associated with HCM pathogenicity with 100% and 94% specificity, respectively. Furthermore, we uncovered that 11% of nontruncating MYBPC3 variants currently classified as of uncertain significance in ClinVar induced one of these molecular phenotypes. Our strategy, which can be applied to other conditions induced by protein loss of function, supports the idea that cMyBP-C haploinsufficiency is a fundamental pathomechanism in HCM.
Keywords: CD; alternative splicing; bioinformatics; cardiac myosin-binding protein C; hypertrophic cardiomyopathy; minigene; protein stability; variants of uncertain significance.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest L. M. holds share in Health in Code. All other authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
The mechanics of the heart: zooming in on hypertrophic cardiomyopathy and cMyBP-C.FEBS Lett. 2022 Mar;596(6):703-746. doi: 10.1002/1873-3468.14301. Epub 2022 Feb 28. FEBS Lett. 2022. PMID: 35224729 Review.
-
Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice.J Mol Cell Cardiol. 2015 Feb;79:234-43. doi: 10.1016/j.yjmcc.2014.11.018. Epub 2014 Nov 25. J Mol Cell Cardiol. 2015. PMID: 25463273 Free PMC article.
-
Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy.Pflugers Arch. 2019 May;471(5):781-793. doi: 10.1007/s00424-018-2226-9. Epub 2018 Nov 20. Pflugers Arch. 2019. PMID: 30456444 Free PMC article. Review.
-
Comparison of the effects of a truncating and a missense MYBPC3 mutation on contractile parameters of engineered heart tissue.J Mol Cell Cardiol. 2016 Aug;97:82-92. doi: 10.1016/j.yjmcc.2016.03.003. Epub 2016 Apr 22. J Mol Cell Cardiol. 2016. PMID: 27108529
-
A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay.Circulation. 2019 Feb 5;139(6):799-811. doi: 10.1161/CIRCULATIONAHA.118.034624. Circulation. 2019. PMID: 30586709 Free PMC article.
Cited by
-
Case report: Severe arrhythmogenic cardiomyopathy in a young girl with compound heterozygous DSG2 and MYBPC3 variants with a 6-year follow-up.Front Genet. 2025 Mar 6;16:1545561. doi: 10.3389/fgene.2025.1545561. eCollection 2025. Front Genet. 2025. PMID: 40115818 Free PMC article.
-
Molecular Epidemiology of Mitochondrial Cardiomyopathy: A Search Among Mitochondrial and Nuclear Genes.Int J Mol Sci. 2021 May 27;22(11):5742. doi: 10.3390/ijms22115742. Int J Mol Sci. 2021. PMID: 34072184 Free PMC article. Review.
-
Integrative single-cell RNA-seq and ATAC-seq analysis of myogenic differentiation in pig.BMC Biol. 2023 Feb 1;21(1):19. doi: 10.1186/s12915-023-01519-z. BMC Biol. 2023. PMID: 36726129 Free PMC article.
-
Protein nanomechanics in biological context.Biophys Rev. 2021 Aug 7;13(4):435-454. doi: 10.1007/s12551-021-00822-9. eCollection 2021 Aug. Biophys Rev. 2021. PMID: 34466164 Free PMC article. Review.
-
Myosin folding boosts solubility in cardiac muscle sarcomeres.JCI Insight. 2024 Mar 14;9(8):e178131. doi: 10.1172/jci.insight.178131. JCI Insight. 2024. PMID: 38483507 Free PMC article.
References
-
- Braunwald E. Hypertrophic cardiomyopathy: The past, the present, and the future. In: Naidu S.S., editor. Hypertrophic Cardiomyopathy. Springer-Verlag; London, UK: 2015. pp. 1–8.
-
- Semsarian C., Ingles J., Maron M.S., Maron B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015;65:1249–1254. - PubMed
-
- Harper A.R., Goel A., Grace C., Thomson K.L., Petersen S.E., Xu X., Waring A., Ormondroyd E., Kramer C.M., Ho C.Y., Neubauer S., Kolm P., Kwong R., Dolman S.F., Desvigne-Nickens P. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021;53:135–142. - PMC - PubMed
-
- Semsarian C., Ingles J. Genetics of HCM and role of genetic testing. In: Naidu S.S., editor. Hypertrophic Cardiomyopathy. Springer-Verlag; London, UK: 2015.
-
- MacArthur D.G., Manolio T.A., Dimmock D.P., Rehm H.L., Shendure J., Abecasis G.R., Adams D.R., Altman R.B., Antonarakis S.E., Ashley E.A., Barrett J.C., Biesecker L.G., Conrad D.F., Cooper G.M., Cox N.J. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
