

ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy
- PMID: 34099552
- PMCID: PMC8214690
- DOI: 10.1073/pnas.2025053118
ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy
Abstract
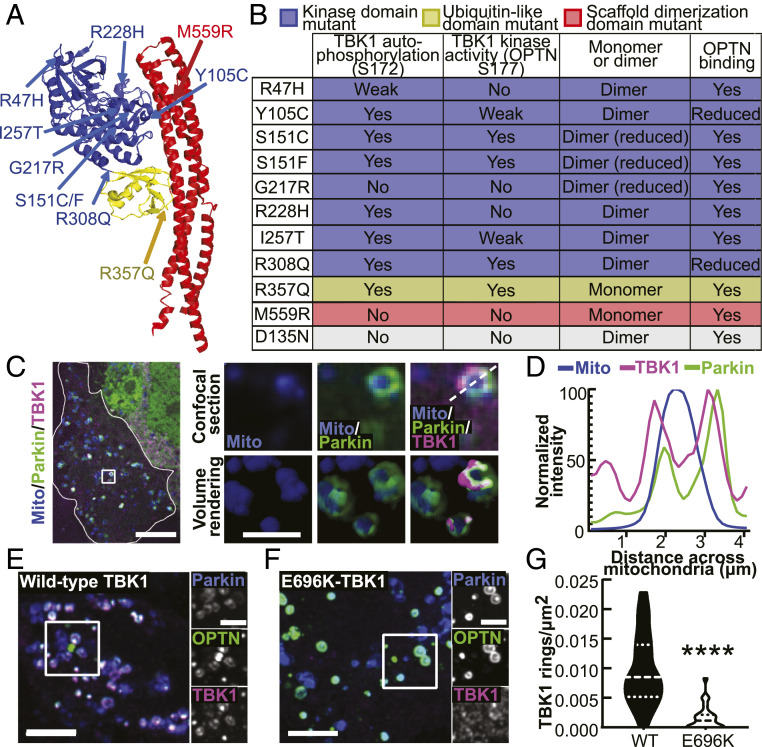
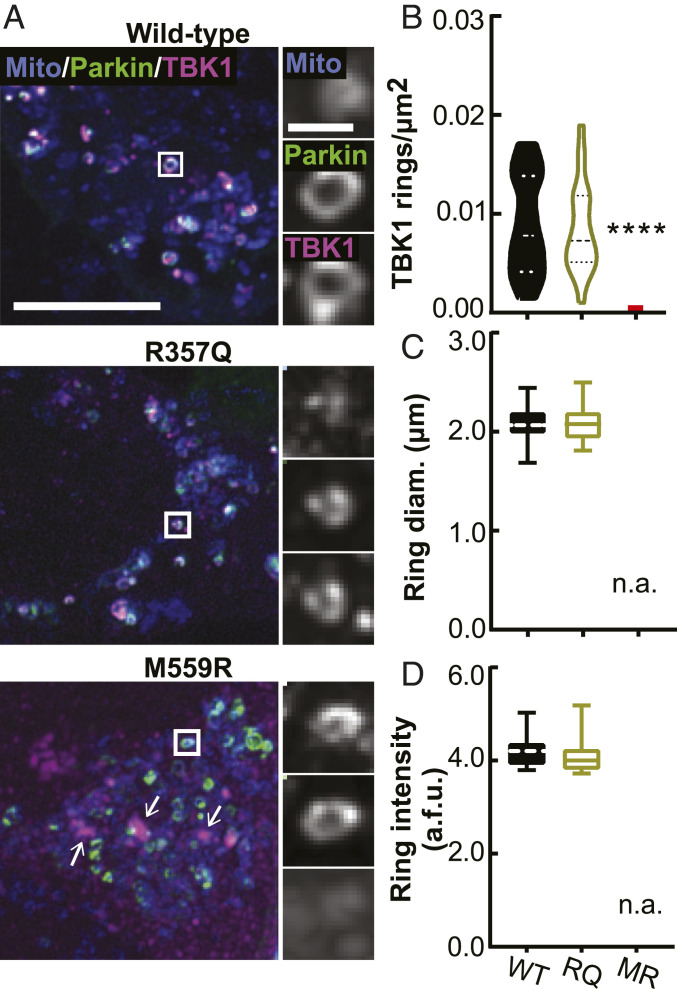
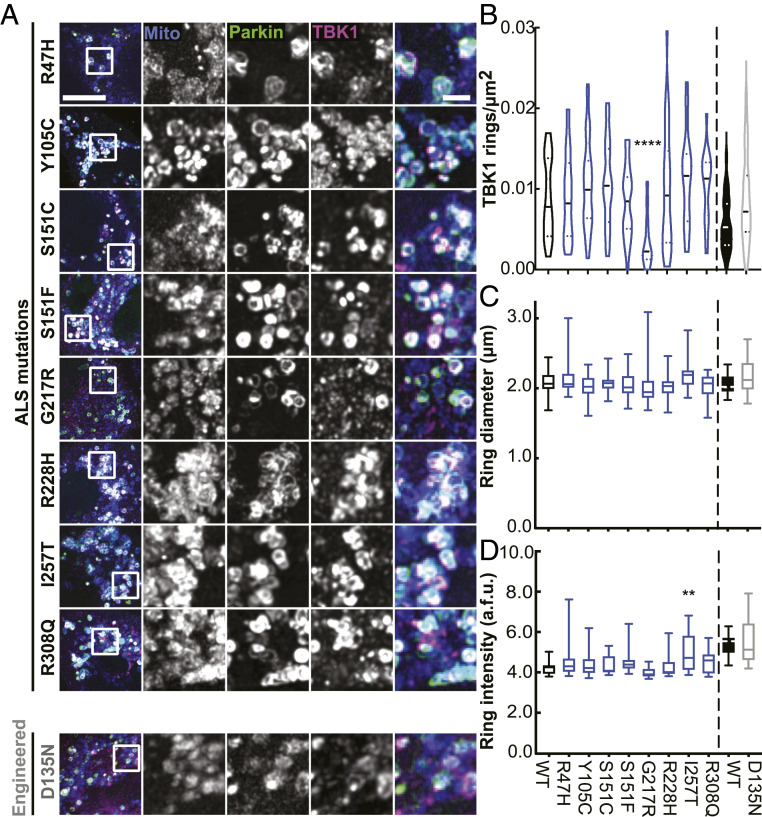
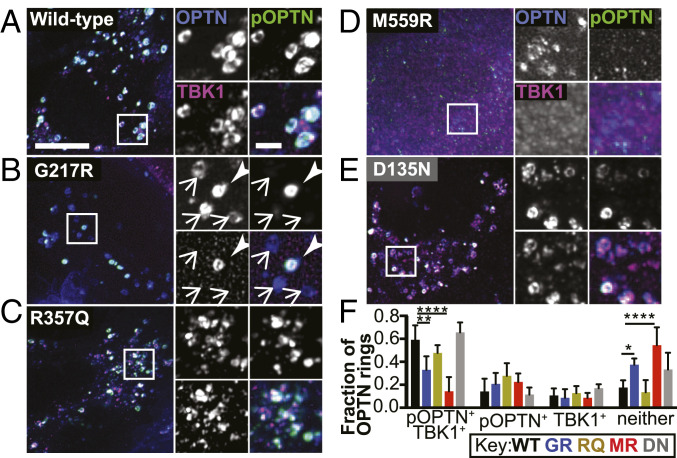
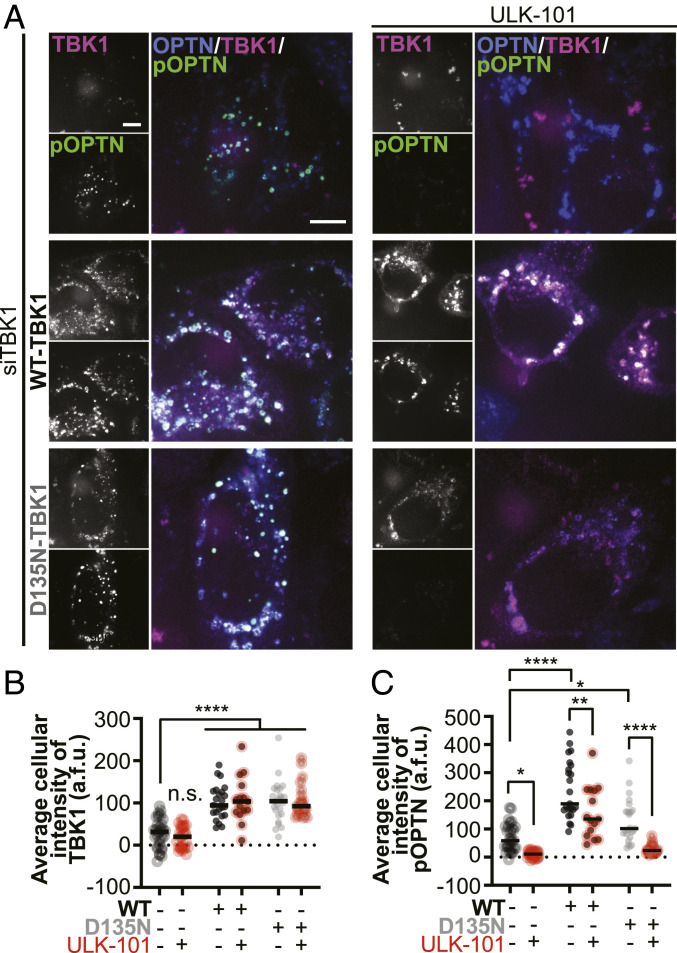
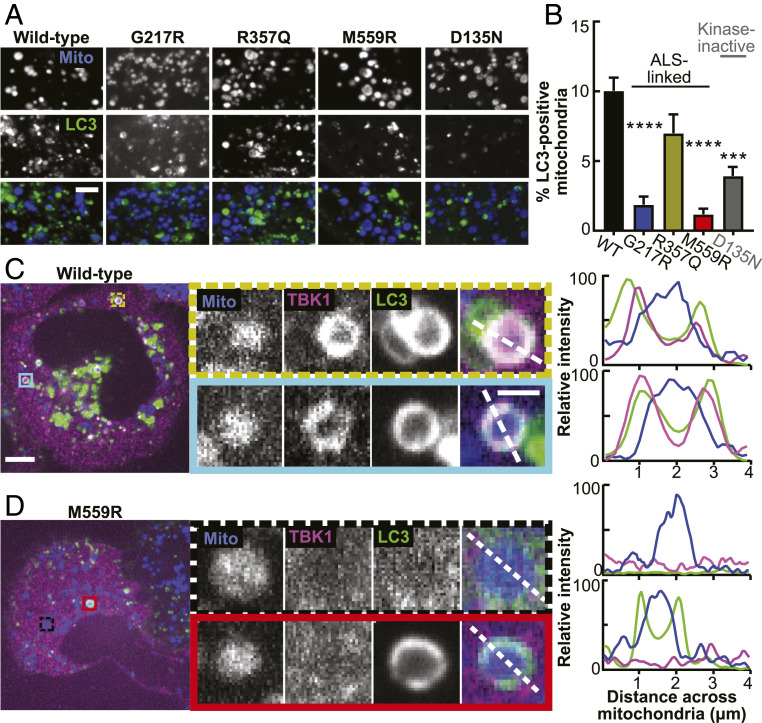
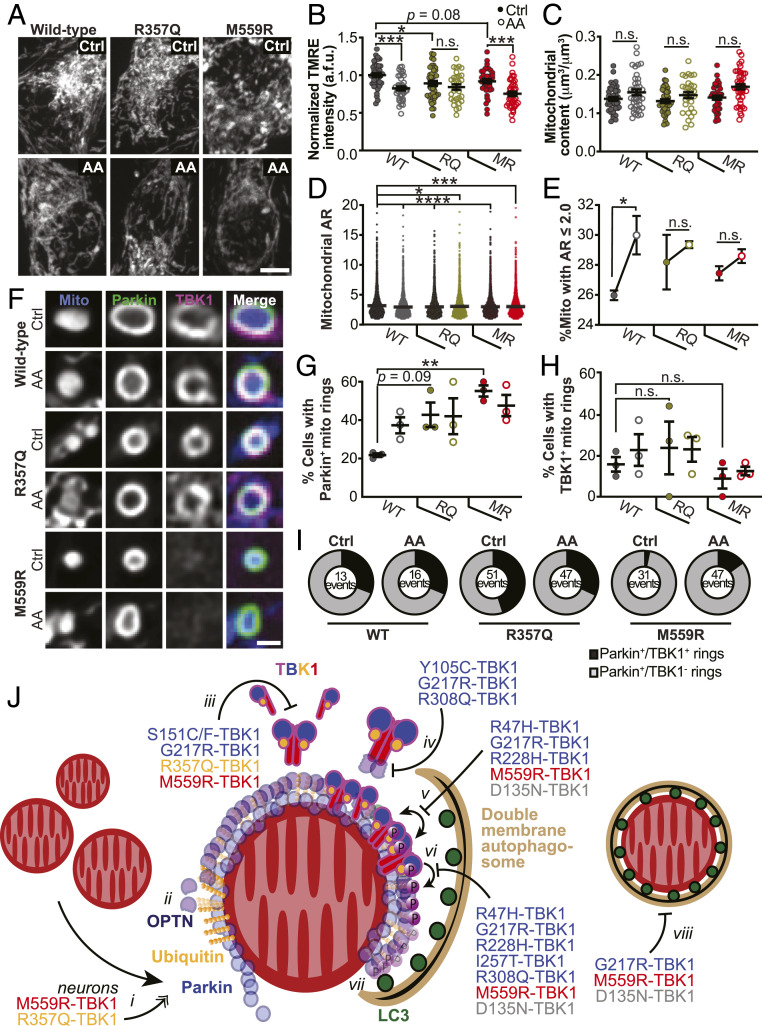
TANK-binding kinase 1 (TBK1) is a multifunctional kinase with an essential role in mitophagy, the selective clearance of damaged mitochondria. More than 90 distinct mutations in TBK1 are linked to amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia, including missense mutations that disrupt the abilities of TBK1 to dimerize, associate with the mitophagy receptor optineurin (OPTN), autoactivate, or catalyze phosphorylation. We investigated how ALS-associated mutations in TBK1 affect Parkin-dependent mitophagy using imaging to dissect the molecular mechanisms involved in clearing damaged mitochondria. Some mutations cause severe dysregulation of the pathway, while others induce limited disruption. Mutations that abolish either TBK1 dimerization or kinase activity were insufficient to fully inhibit mitophagy, while mutations that reduced both dimerization and kinase activity were more disruptive. Ultimately, both TBK1 recruitment and OPTN phosphorylation at S177 are necessary for engulfment of damaged mitochondra by autophagosomal membranes. Surprisingly, we find that ULK1 activity contributes to the phosphorylation of OPTN in the presence of either wild-type or kinase-inactive TBK1. In primary neurons, TBK1 mutants induce mitochondrial stress under basal conditions; network stress is exacerbated with further mitochondrial insult. Our study further refines the model for TBK1 function in mitophagy, demonstrating that some ALS-linked mutations likely contribute to disease pathogenesis by inducing mitochondrial stress or inhibiting mitophagic flux. Other TBK1 mutations exhibited much less impact on mitophagy in our assays, suggesting that cell-type-specific effects, cumulative damage, or alternative TBK1-dependent pathways such as innate immunity and inflammation also factor into the development of ALS in affected individuals.
Keywords: OPTN; Parkin; TBK1; mitophagy; neurodegeneration.
Copyright © 2021 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures

Similar articles
-
Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy.Proc Natl Acad Sci U S A. 2016 Jun 14;113(24):E3349-58. doi: 10.1073/pnas.1523810113. Epub 2016 May 31. Proc Natl Acad Sci U S A. 2016. PMID: 27247382 Free PMC article.
-
Frustration analysis of TBK1 missense mutations reported in ALS/FTD and cancer patients.3 Biotech. 2022 Aug;12(8):174. doi: 10.1007/s13205-022-03240-0. Epub 2022 Jul 14. 3 Biotech. 2022. PMID: 35845111 Free PMC article.
-
Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis.Neurobiol Dis. 2022 Nov;174:105859. doi: 10.1016/j.nbd.2022.105859. Epub 2022 Sep 13. Neurobiol Dis. 2022. PMID: 36113750 Review.
-
Effects of ALS-associated TANK binding kinase 1 mutations on protein-protein interactions and kinase activity.Proc Natl Acad Sci U S A. 2019 Dec 3;116(49):24517-24526. doi: 10.1073/pnas.1915732116. Epub 2019 Nov 20. Proc Natl Acad Sci U S A. 2019. PMID: 31748271 Free PMC article.
-
TBK1: a new player in ALS linking autophagy and neuroinflammation.Mol Brain. 2017 Feb 2;10(1):5. doi: 10.1186/s13041-017-0287-x. Mol Brain. 2017. PMID: 28148298 Free PMC article. Review.
Cited by
-
SRF deletion results in earlier disease onset in a mouse model of amyotrophic lateral sclerosis.JCI Insight. 2023 Aug 8;8(15):e167694. doi: 10.1172/jci.insight.167694. JCI Insight. 2023. PMID: 37339001 Free PMC article.
-
RCHY1 and OPTN are required for melanophagy, selective autophagy of melanosomes.Proc Natl Acad Sci U S A. 2024 Apr 2;121(14):e2318039121. doi: 10.1073/pnas.2318039121. Epub 2024 Mar 27. Proc Natl Acad Sci U S A. 2024. PMID: 38536750 Free PMC article.
-
Mitochondrial damage triggers the concerted degradation of negative regulators of neuronal autophagy.Nat Commun. 2025 Aug 9;16(1):7367. doi: 10.1038/s41467-025-62379-5. Nat Commun. 2025. PMID: 40783388 Free PMC article.
-
Loss-of-function variants in RNA binding motif protein X-linked induce neuronal defects contributing to amyotrophic lateral sclerosis pathogenesis.MedComm (2020). 2024 Sep 10;5(9):e712. doi: 10.1002/mco2.712. eCollection 2024 Sep. MedComm (2020). 2024. PMID: 39263607 Free PMC article.
-
Optineurin provides a mitophagy contact site for TBK1 activation.EMBO J. 2024 Mar;43(5):754-779. doi: 10.1038/s44318-024-00036-1. Epub 2024 Jan 29. EMBO J. 2024. PMID: 38287189 Free PMC article.
References
-
- Freischmidt A., et al. ., Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015). - PubMed
-
- Freischmidt A., Müller K., Ludolph A. C., Weishaupt J. H., Andersen P. M., Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol. 74, 110–113 (2017). - PubMed
-
- van Rheenen W.et al. .; PARALS Registry; SLALOM Group; SLAP Registry; FALS Sequencing Consortium; SLAGEN Consortium; NNIPPS Study Group , Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 48, 1043–1048 (2016). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
