

High throughput screening identifies SOX2 as a super pioneer factor that inhibits DNA methylation maintenance at its binding sites
- PMID: 34099689
- PMCID: PMC8184831
- DOI: 10.1038/s41467-021-23630-x
High throughput screening identifies SOX2 as a super pioneer factor that inhibits DNA methylation maintenance at its binding sites
Abstract
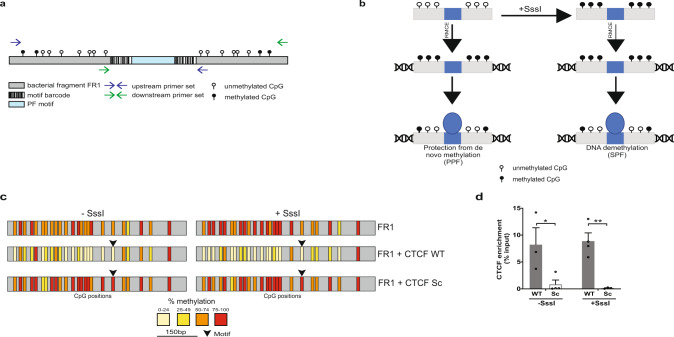
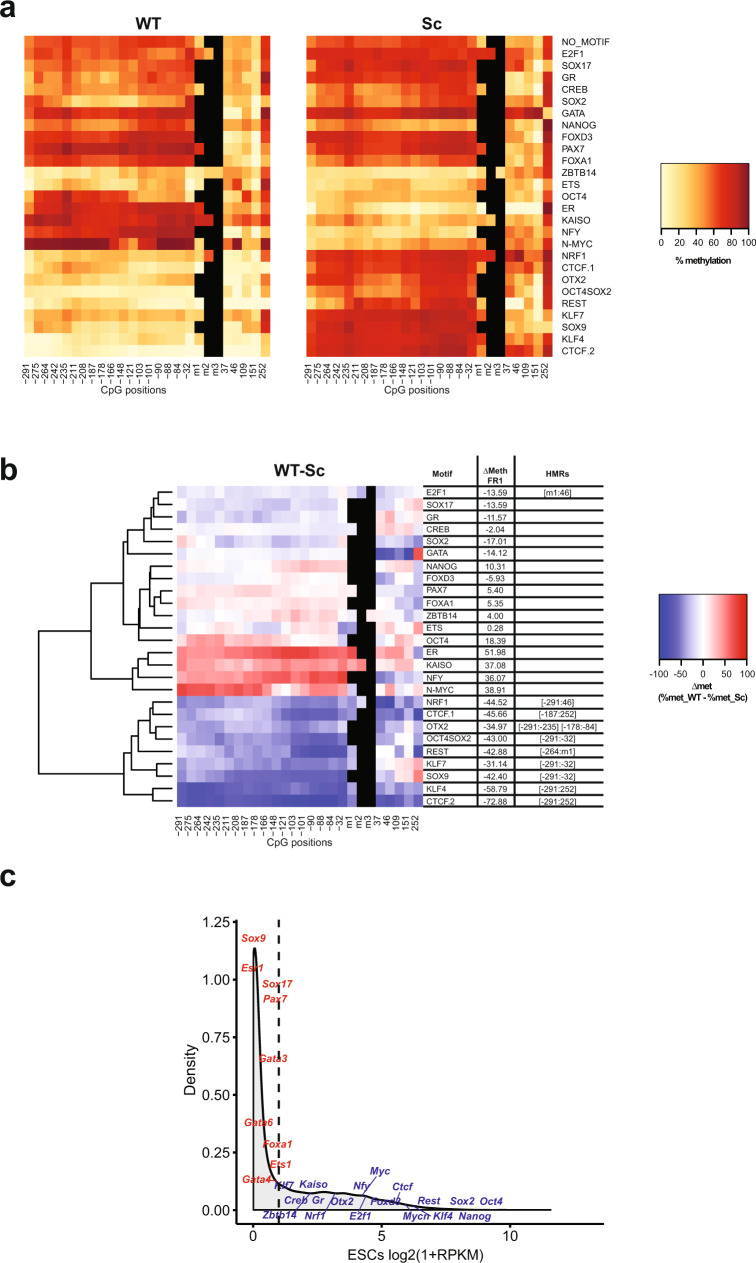
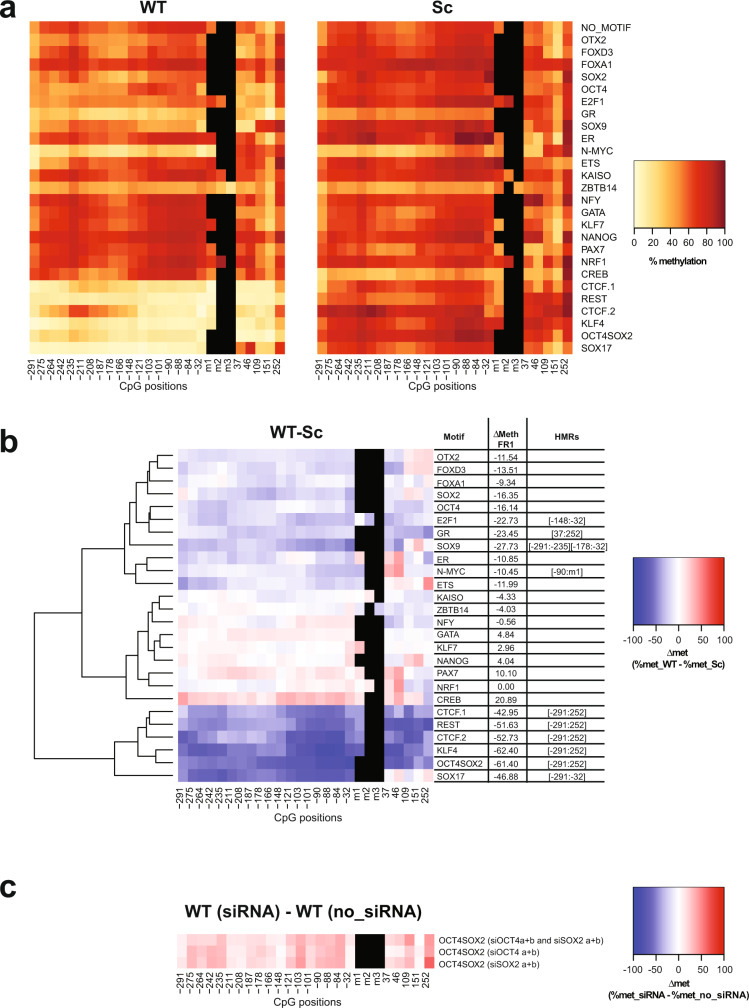
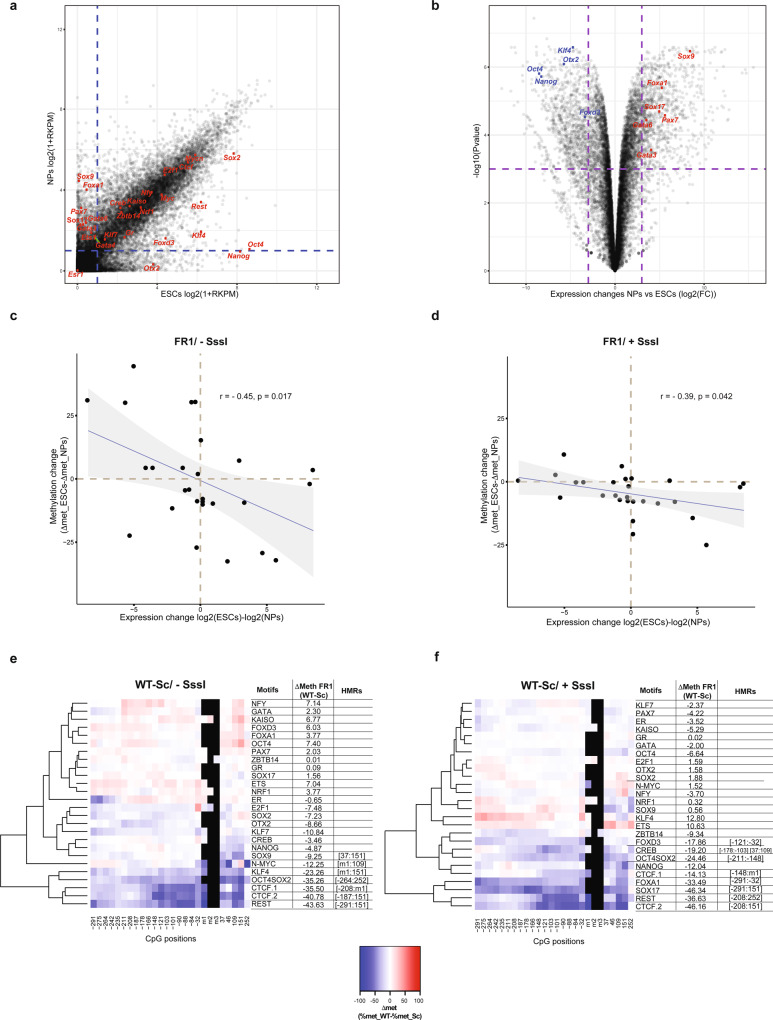
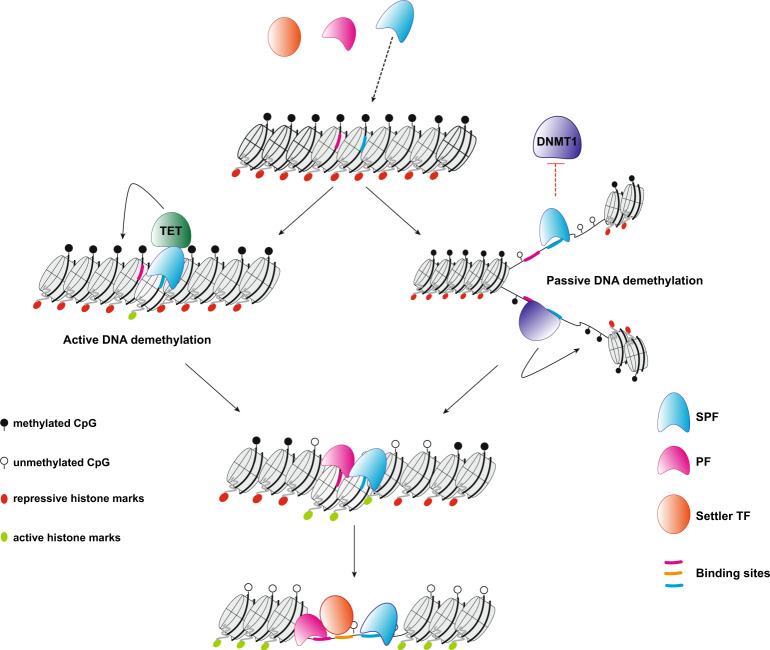
Binding of mammalian transcription factors (TFs) to regulatory regions is hindered by chromatin compaction and DNA methylation of their binding sites. Nevertheless, pioneer transcription factors (PFs), a distinct class of TFs, have the ability to access nucleosomal DNA, leading to nucleosome remodelling and enhanced chromatin accessibility. Whether PFs can bind to methylated sites and induce DNA demethylation is largely unknown. Using a highly parallelized approach to investigate PF ability to bind methylated DNA and induce DNA demethylation, we show that the interdependence between DNA methylation and TF binding is more complex than previously thought, even within a select group of TFs displaying pioneering activity; while some PFs do not affect the methylation status of their binding sites, we identified PFs that can protect DNA from methylation and others that can induce DNA demethylation at methylated binding sites. We call the latter super pioneer transcription factors (SPFs), as they are seemingly able to overcome several types of repressive epigenetic marks. Finally, while most SPFs induce TET-dependent active DNA demethylation, SOX2 binding leads to passive demethylation, an activity enhanced by the co-binding of OCT4. This finding suggests that SPFs could interfere with epigenetic memory during DNA replication.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Genome access is transcription factor-specific and defined by nucleosome position.Mol Cell. 2024 Sep 19;84(18):3455-3468.e6. doi: 10.1016/j.molcel.2024.08.009. Epub 2024 Aug 28. Mol Cell. 2024. PMID: 39208807 Free PMC article.
-
Structural Plasticity of Pioneer Factor Sox2 and DNA Bendability Modulate Nucleosome Engagement and Sox2-Oct4 Synergism.J Mol Biol. 2023 Jan 30;435(2):167916. doi: 10.1016/j.jmb.2022.167916. Epub 2022 Dec 7. J Mol Biol. 2023. PMID: 36495920 Free PMC article.
-
Nonreciprocal and Conditional Cooperativity Directs the Pioneer Activity of Pluripotency Transcription Factors.Cell Rep. 2019 Sep 3;28(10):2689-2703.e4. doi: 10.1016/j.celrep.2019.07.103. Cell Rep. 2019. PMID: 31484078 Free PMC article.
-
Changing the DNA landscape: putting a SPN on chromatin.Curr Top Microbiol Immunol. 2003;274:171-201. doi: 10.1007/978-3-642-55747-7_7. Curr Top Microbiol Immunol. 2003. PMID: 12596908 Review.
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
Cited by
-
The homeodomain of Oct4 is a dimeric binder of methylated CpG elements.Nucleic Acids Res. 2023 Feb 22;51(3):1120-1138. doi: 10.1093/nar/gkac1262. Nucleic Acids Res. 2023. PMID: 36631980 Free PMC article.
-
A bipartite element with allele-specific functions safeguards DNA methylation imprints at the Dlk1-Dio3 locus.Dev Cell. 2021 Nov 22;56(22):3052-3065.e5. doi: 10.1016/j.devcel.2021.10.004. Epub 2021 Oct 27. Dev Cell. 2021. PMID: 34710357 Free PMC article.
-
Phospho-seq: integrated, multi-modal profiling of intracellular protein dynamics in single cells.Nat Commun. 2025 Feb 4;16(1):1346. doi: 10.1038/s41467-025-56590-7. Nat Commun. 2025. PMID: 39905064 Free PMC article.
-
Active DNA demethylation of developmental cis-regulatory regions predates vertebrate origins.Sci Adv. 2022 Dec 2;8(48):eabn2258. doi: 10.1126/sciadv.abn2258. Epub 2022 Dec 2. Sci Adv. 2022. PMID: 36459547 Free PMC article.
-
Enhancer methylation dynamics drive core transcriptional regulatory circuitry in pan-cancer.Oncogene. 2022 Jun;41(26):3474-3484. doi: 10.1038/s41388-022-02359-x. Epub 2022 Jun 2. Oncogene. 2022. PMID: 35655092
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
